
| Introduction | Top of page |
Due to shared modes of transmission, coinfection with hepatitis B virus (HBV) and HIV is common. With a reduction in AIDS-related deaths due to highly active antiretroviral therapy (HAART), liver disease has emerged as an important cause of death in patients with HBV-HIV coinfection.
Hepatitis B is a dynamic disease and an understanding of its virology and natural history is imperative if complications are to be reduced and disease progression limited. The management of HBV-HIV coinfection is complicated by the use of drugs with activity against both viruses, the risk of flares and hepatic decompensation with immune reconstitution, and the increasing prevalence of antiviral resistance.
| Epidemiology | Top of page |
More than 350 million people are infected with HBV, with 75% of the world’s HBV carriers residing in Asia (Lee, 1997; Lai, 2003; Burnett, 2005). Forty million people are infected with HIV worldwide. Due to shared modes of transmission, coinfection is common, and an estimated 4 million people worldwide are coinfected with HBV-HIV. The prevalence of HBV in HIV-infected individuals varies with the population studied. In the United States, up to 10% of all HIV-infected individuals have HBV coinfection (Thio, 2003). Several studies support an increased prevalence of HBV in HIV-infected populations of sub-Saharan Africa, with more than 80% of HIV-positive individuals in some of those countries carrying serum markers for HBV (Burnett, 2005).
Figure 1.Geographic Distribution of Chronic HBV Infection.
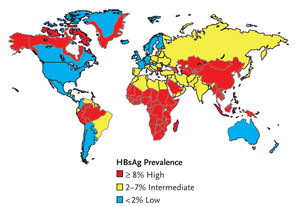
HBsAg, hepatitis B surface antigen Source: Department of Health and Human Services Centers for Disease Control and Prevention. Viral Hepatitis Slidesets: Hepatitis B 101. Available here. Accessed January 15, 2007.
The prevalence of HBV surface antigen (HBsAg) varies geographically, with high endemicity areas (≥8%) in Southeast Asia, sub-Saharan Africa, the Amazon Basin, parts of the Middle East, the central Asian republics, and parts of Eastern Europe. Areas of low endemicity (<2%) include North America, Western and Northern Europe, Australia, and parts of South America (Figure 1) (WHO, 2002).
Hepatitis B genotypes (A-H) geographically vary in distribution. Genotypes B and C are prevalent in Southeast Asia (Enomoto, 2006). Genotype A predominates in sub-Saharan Africa (genotype Aa) and Europe (genotype Ae). Genotype F is most common in South America. All 8 known genotypes are found in North America because of immigration from affected areas.
The HBV genotype is increasingly being recognized for its role in disease progression and response to therapy (Buti, 2005); however, there is no role for determining genotype in all HBV-infected individuals, particularly those with HIV coinfection. Genotype A is the predominant genotype in HBV-HIV coinfected individuals in the United States and Europe (Lacombe, 2006). Genotype A is more responsive to interferon than genotype D, while genotype B is more responsive to interferon than genotype C (Enomoto, 2006). Clearance of HBsAg has been shown to occur more often with genotype A compared with genotype D. Genotype B is associated with less-active and more slowly progressive liver disease compared with genotype C. Genotype D is associated with the precore and core promoter mutations and, therefore, usually necessitates long-term therapy (Buti, 2005).
| Hepatitis B Virology | Top of page |
The HBV genome is a circular, partially double-stranded DNA with 4 overlapping reading frames encoding the envelope, core, polymerase, and X proteins (Lee, 1997; Lok, 2001). The envelope protein is found in serum as surface antigen (HBsAg). The gene encoding core antigen (HBcAg) also encodes e antigen (HBeAg) using an upstream start site. The HBeAg contains a signal peptide that targets it to the endoplasmic reticulum for secretion into serum, while HBcAg does not contain a signal peptide and is incorporated into the virion. A mutation between the start sites of HBeAg and HBcAg (precore or core promoter region) decreases or abolishes the production of HBeAg without affecting the production of HBcAg. The prevalence of these mutations increases over time and is, therefore, more common in individuals who acquired HBV perinatally or at a young age. Although the hepatitis B virion itself does not cross the placental barrier, HBeAg can cross the placenta and may function as an immune desensitizer to HBV, possibly predisposing the fetus to the establishment of chronic HBV infection. The polymerase protein has both reverse transcriptase and DNA polymerase activity. The X protein may play a role in the development of hepatocellular carcinoma (HCC) (Lee, 1997; Lok, 2001).
Hepatitis B virus is not directly cytopathic. The spectrum of disease in HBV is determined by the host immune response, where hepatocyte injury leading to acute and chronic hepatitis is mediated by cytotoxic T lymphocytes (Fukuda, 1995; Lee, 1997). The profound immune response in acute HBV infection and the aminotransferase elevations accompanying HBeAg seroconversion are a response to the immune-mediated clearance of infected hepatocytes.
| Table 1. Initial Evaluation of a Patient with HIV and Newly Diagnosed HBV. | History | Alcohol useFamily history of hepatocellularcarcinomaLength of hbv infection(childhood or as an adult) |
| Physical Exam | JaundiceSplenomegalyGynecomastiaSpider angiomataPalmar erythemaAsterixis |
| Hepatic Synthetic Function | Prothrombin timeSerum albumin |
| Markers of Inflammation/Fibrosis/Portal hypertension | HBeAganti-HBehbv dna |
| Markers of hbv Replication | H BeAganti-HBehbv dna |
| Tests for Coinfection | Anti-hav (total): vaccinate ifnegativeAnti-hcvConsider anti-hdv |
| Screen for hcc | Alpha fetoproteinImaging study |
| alt, alanine aminotransferase; anti-HBe, antibody to hepatitis B e antigen; ast, aspartate aminotransferase; cbc, complete blood count; hav, hepatitis A virus; hbv, hepatitis B virus; HBeAg, hepatitis B e antigen; hcc, hepatocellular carcinoma; hcv, hepatitis C virus; hdv, hepatitis D virus | |
| Risk Factors | Top of page |
The most common risk factors for acquiring HBV as an adult are injection drug use and sexual contact, particularly with multiple sexual partners and men who have sex with men. In one-third of cases, a cause cannot be identified, possibly because of a reluctance to report high-risk behavior, or unrecognized modes of transmission (Lee, 1997). Although risk factors are similar, HBV is more efficiently transmitted than HIV: percutaneous exposure to infected blood carries a 30% risk of HBV transmission compared with a 0.3% risk of HIV transmission.
| Screening And Vaccination | Top of page |
All HIV-positive patients should be screened for HBV with HBsAg, hepatitis B surface antibody (anti-HBs) and total hepatitis B core antibody (anti-HBc). If these tests are negative, the patient should receive HBV vaccination, though HIV-infected patients do not respond as well to HBV vaccination as HIV-uninfected patients, and the persistence of protective surface antibody is short-lived (Rey, 2000). The response is poorer in patients with CD4 cell counts between 200 and 500 cells/μL compared with patients with CD4 cell counts above 500 cells/μL, with response rates of 33% and 87.5%, respectively (Rey, 2000). Patients who fail to respond to a conventional course of vaccine (20 μg) should receive booster doses or a repeat cycle with the 40-μg dose (Brook, 2005).
If any of these tests (HBsAg, anti-HBs, anti-HBc) are positive, HBV DNA should be measured, since atypical serologies occur in HIV coinfection (see “Occult Infection” below). Household and sexual contacts should also be screened and vaccinated if not already immune.
| Initial Evaluation | Top of page |
The initial evaluation of a patient with HIV and newly identified HBV should include a history and physical examination, with attention to alcohol use and family history of HBV and HCC. Laboratory tests should assess liver disease (liver enzymes, complete blood count (CBC) with platelets and prothrombin time), markers of HBV replication (HBeAg and anti-HBe, HBV DNA), and tests for coinfection, including antibody to hepatitis D virus (HDV) in high-risk groups (Mediterranean and parts of South America) and antibody to hepatitis C virus (HCV) (Lok, 2001; Brook, 2005). Total antibody to hepatitis A virus (HAV) should be checked and, if negative, the patient should receive 2 doses of HAV vaccine 6 to 12 months apart (Brook, 2005). All patients should be screened for HCC with serum alpha fetoprotein and liver ultrasonography (Lok, 2001). Liver biopsy should be considered in patients with HBV replication, especially with abnormal liver enzymes and in HDV or HCV coinfection (Table 1) (Brook, 2005).
| Natural History | Top of page |
After acute infection, there is an early replicative phase of HBV. It is during this phase that patients are most infectious and at greatest risk for development of progressive liver disease (Davis, 1991). The risk of developing chronic HBV following infection varies with age of HBV acquisition (Table 2). Perinatal acquisition and infection in children younger than 1 year leads to chronic infection in 90% to 100% of cases, whereas less than 5% of adults who acquire HBV develop chronic infection (Lai, 2003). Individuals with HIV are at significantly higher risk of developing chronic HBV, with a risk of 21% in unvaccinated persons (Hadler, 1991).
In chronic infection, HBV DNA titers decline over time, though HBsAg remains detectable. There is a spontaneous loss of HBeAg and anti-HBe seroconversion at a rate of about 5% to 10% per year, leading to a phase of nonreplication or low-level replication (Davis, 1991; Ganem, 2004). A flare, or transient rise in alanine aminotransferase (ALT) levels, often accompanies this seroconversion (Ganem, 2004). What follows is a period of low HBV DNA levels, detectable only by polymerase chain reaction, and normal ALT levels (Davis, 1991). Spontaneous loss of HBsAg and anti-HBs seroconversion is less common, with an annual incidence below 1% (Fattovich, 1998; Niederau, 1996).
| Table 2. Risk of Chronic HBV by Age of Acquisition and Immune Status. | |
| Neonates | 90%–100% |
| Children | 20%–40% |
| hiv-positive | 21% (Hadler, 1991) |
| Adults | <5% |
| hbv, hepatitis B virus; hiv, human immunodeficiency virus | |
It is important to understand that seroconversion (loss of HBeAg or HBsAg and development of anti-HBe or anti-HBs) does not imply permanent remission of chronic HBV. A proportion of these patients can reactivate, with reappearance of HBV DNA, HBeAg, and elevation of ALT levels. Spontaneous reactivation is an important cause of progression of liver injury (Davis, 1984). Additionally, immune suppression, particularly following corticosteroid use or chemotherapy, are known causes of reactivation (Hui, 2006). Even patients who are anti-HBc positive but HBsAg negative are at risk for reactivation (Brook, 2005).
In a longitudinal study of children with chronic HBV, of 89 children who developed anti-HBe seroconversion during the study period, 4 had already developed cirrhosis, and 2 developed HCC. Of the 85 children without cirrhosis, 4 untreated children reactivated during the study period (Bortolotti, 2006). This underscores the importance of continuous monitoring of patients with chronic HBV following anti-HBe seroconversion (see “Monitoring” below). Hepatitis B markers (HBV DNA, HBeAg and anti-HBe) should be checked annually or if ALT becomes abnormal (Brook, 2005).
Cirrhosis develops in 30% of individuals who are chronically infected with HBV (Torresi, 2000). Compared with HBV-uninfected individuals, the risk of HCC is increased 100-fold in those who are HBsAg positive, of whom 5% to 10% develop HCC (Torresi, 2000). Approximately 23% of patients with HBV develop hepatic decompensation (ascites, jaundice, hepatic encephalopathy or variceal bleeding) within 5 years of developing cirrhosis (Fattovich, 1995). After the first episode of decompensation, the risk of death within 5 years is 65% (Fattovich, 1995). Patients with chronic HBV have a 25%-40% lifetime risk of death from liver failure or HCC (Perrillo, 2001). Chronic HBV is the sixth most common indication for liver transplantation for adults in the United States (Perrillo, 2001). Although HIV was once a contraindication to solid organ transplantation, liver transplantation of HBV-HIV coinfected patients is now performed in a limited number of centers (Roland, 2006).
| Impact Of HIV on Hepatitis B | Top of page |
| Table 3. Impact of HIV on HBV. | |
| Prolonged alt elevation | |
| Reduced rate of spontaneous HBeAg and HBsAg seroconversion | |
| Increased rate of HBeAg-positive disease | |
| Higher hbv dna levels | |
| Lower alt elevations | |
| Milder hepatic necroinflammation | |
| Increased progression to cirrhosis | |
| Increased risk of hcc | |
| Decreased response to interferon | |
| Decreased efficacy of anti-hbv therapy | |
| Increased lamivudine-resistant mutations | |
| alt, alanine aminotransferase; HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; hbv, hepatitis B virus; hcc, hepatocellular carcinoma; hiv, human immunodeficiency virus; 3tc, lamivudine | |
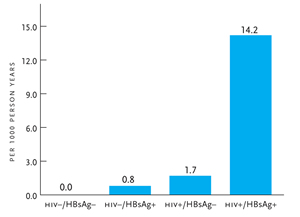
Figure 2. Liver-Related Mortality due to HIV and HBV.
Both HIV-1 and HIV-2 are of primate origin. The origin of HIV-2 has been established to be the sooty mangabey, an Old World monkey of Guinea Bissau, Gabon, and Cameroon. The origin of HIV-1 is the central subspecies of chimpanzee, pictured here.
HBsAg, hepatitis B surface antigen; HIV, human immunodeficiency virus Reprinted with permission from Thio CL, et al. Lancet. 2002;360:1921–1926.
As shown in Table 3, HIV has a significant impact on the natural history of HBV infection. The presence of HIV prior to HBV infection increases the risk of developing chronic HBV and prolonged ALT elevation (Hadler, 1991). Hepatitis B-HIV coinfection reduces the rate of spontaneous HBeAg and HBsAg seroconversion, leading to a higher prevalence of HBeAg-positive disease (Thio, 2003). There is an association between HIV and reactivation of HBV and elevated HBV DNA levels, although serum ALT elevations are milder compared with HBV monoinfected individuals (Colin, 1999; Thio, 2003). Despite this, liver damage progresses more rapidly, condensing the period from HBV acquisition to cirrhosis in individuals with HBV-HIV coinfection (Thio, 2003). Coinfected patients have a poorer response to interferon therapy.
Patients with HBV-HIV coinfection have an increased risk of liver-related complications and death. Since the introduction of HAART in 1996, a major reduction in the incidence of opportunistic infections has led to the emergence of liver disease as one of the leading causes of death in patients with HIV. In a retrospective review, 50% of deaths in a cohort of HIV-infected individuals in 1998 through 1999 were due to end-stage liver disease compared with less than 14% of deaths in 1991 and 1996 (Bica, 2001). A large cohort study showed that the liver-related mortality rate in those who were HBV-HIV coinfected was 14.2 per 1000 person-years, compared with 0.8 per 1000 person-years for HBV monoinfection and 1.7 per 1000 person-years for HIV alone (Figure 2) (Thio, 2002). In a more recent prospective study of HIV-infected individuals in the United States, Europe and Australia, liver-related disease was the second most common cause of death after AIDS (14.5% and 31.1%, respectively) (Weber, 2006). Liver-related mortality was more common than death from cardiovascular diseases (9.4%) and non-AIDS malignancies (11%). Active HBV was an independent predictor of liver-related death, with an adjusted relative risk of 3.73.
| Occult Infection | Top of page |
Occult HBV infection is defined as the presence of HBV DNA in the absence of HBsAg (Shire, 2004; Mphahlele, 2006). Often the only serum marker in occult HBV is anti-HBc (Hofer, 1998; Shire, 2004). The prevalence of occult HBV in HIV-infected populations varies from 10% to 43% depending on the study (Hofer, 1998; Shire, 2004; Mphahlele, 2006), whereas the presence of anti-HBc alone is rarely associated with occult infection in HIV-uninfected patients. One study showed ongoing necroinflammatory liver disease with chronic ALT elevations in HIV-infected patients with occult HBV, illustrating that occult HBV represents chronic HBV with active viral replication and should be managed as such (Hofer, 1998).
| Flares And Immune Reconstitution | Top of page |
Flares result from changes in the balance between the level of HBV replication and the immune response (Drake, 2004). Although spontaneous flares and seroconversion occur in HIV-uninfected individuals, this is rarely seen in HIV-infected individuals without HAART (Carr, 1997). A substantial reduction in HIV viremia and improvement in CD4 cell count after the initiation of HAART, called immune reconstitution, can lead to improved host immune response to HBV and other viral opportunistic infections. In HBV coinfection, immune reconstitution is often manifested by a flare followed by reduction in HBV viremia and less commonly seroconversion. In addition to immune reconstitution, flares of HBV disease in individuals with HIV can occur when anti-HBV therapy is withdrawn, when anti-HBV drug resistance develops (Bessesen, 1999; Pillay, 2000), or because of HAART-related hepatotoxicity (Drake, 2004).
Occasionally, a severe flare associated with immune reconstitution in patients with high-level HBV DNA can cause hepatic decompensation and even death (Bessesen, 1999; Drake, 2004). This experience highlights the importance of screening all HIV-infected patients prior to initiation of HAART, and control of HBV prior to or in conjunction with initiation of HAART (Drake, 2004). Control of HBV with an anti-HBV drug that has no anti-HIV activity prior to initiation of HAART should be considered in patients with advanced fibrosis and cirrhosis who are at higher risk for hepatic decompensation resulting from a flare.
| Other Causes Of Liver Disease | Top of page |
It should be recognized that liver disease in patients with HBV-HIV coinfection can be from causes other than HBV, including other viral infections (HCV, HAV), alcohol, medications, HAART-related hepatotoxicity, and nonalcoholic fatty liver disease (Table 4). Abnormal liver enzymes in HBV-HIV coinfected patients may be due to any of these factors, as well as the development of drug resistance or reactivation of HBV.
| Table 4. Differential Diagnosis of Liver Abnormalities in Patients with HBV-HIV Coinfection. | |
| hbv (flare, reactivation, seroconversion) | |
| Other viral infections (eg, hcv, hdv, hav) | |
| Alcohol | |
| Medications | |
| haart-related hepatotoxicity | |
| Non-alcoholic fatty liver disease | |
| Antiviral resistance | |
| haart, highly active antiretroviral therapy; hav, hepatitis A virus; hbv, hepatitis B virus; hcv, hepatitis C virus; hdv, hepatitis D virus; hiv, human immunodeficiency virus | |
| Therapy | Top of page |
The goals of HBV therapy are to stop or reverse progression of liver inflammation and fibrosis through sustained suppression of HBV replication (Peters, 2006). Studies of HBV therapy use several endpoints, including virologic (decrease in HBV DNA), serologic (anti-HBe or anti-HBs seroconversion), and inflammatory (normalization of ALT or liver histology). It is important to recognize that HBV is probably never cured but, rather, is controlled by limiting viral replication. The benefits of limiting HBV viremia are illustrated by studies that demonstrate a direct association between HBV DNA levels and the risk of developing cirrhosis (Iloeje, 2006) and HCC (Chen, 2006), independent of HBeAg status. Patients who clear HBeAg after interferon therapy have fewer complications and improved overall survival (Niederau, 1996). Loss of HBsAg is associated with the best survival and lowest risk of developing HCC and liver-related death (Fattovich, 1998).
Available oral therapies for HBV-HIV coinfection are shown in Table 5. A brief description of each follows.
| Table 5. Available Oral Anti-HBV Therapies. | |||
| Drug | Treats Wild-Type hbv | Treats 3tc-Resistant (rtM204V/I) hbv | Treats hiv |
| Lamivudine (3tc) | Yes | No | Yes |
| Emtricitabine (ftc) | Yes | No | Yes |
| Adefovir dipivoxil (ADV) | Yes | Yes | No |
| Entecavir (ETV) | Yes (0.5 mg daily) | Yes (1.0 mg daily) | No* |
| Telbivudine (LdT) | Yes | No | No |
| hbv, hepatitis B virus; hiv, human immunodeficiency virus * T he potential for the development of hiv drug resistance while on etv monotherapy for chronic hbv was recently reported in a patient with a prior history of hiv treatment with haart (fda, 2007). |
|||
Lamivudine (3TC), a nucleoside analogue of cytosine, is licensed for the treatment of HIV as well as HBV (at different doses) and is a frequent component of HAART. Resistance to 3TC, most commonly due to mutations in the YMDD motif (eg, rtM204V/I) of the gene encoding the HBV polymerase, develops in up to 90% of HBV-HIV coinfected individuals after 4 years of 3TC therapy, thereby limiting its use as long-term therapy (Benhamou, 1999). Other changes have been described such as rtA181T/V but these are uncommon (Yeh, 2000).
Emtricitabine (FTC), a nucleoside analogue of cytosine similar to 3TC, is approved for the treatment of HIV and has activity against HBV. It shares similar cross-resistance to 3TC (Soriano, 2005).
Adefovir dipivoxil (ADV), a nucleotide analogue of adenosine monophosphate that inhibits HBV polymerase, is active against both wild-type and 3TC-resistant HBV with the rtM204V/I. It is not active against rtA181T/V (see below). Although it is associated with less resistance compared with 3TC, there is still a 29% probability of resistance after 5 years of treatment in HBV-monoinfected individuals (Hadziyannis, 2006).
Entecavir (ETV) is a deoxyguanosine analogue with activity against HBV that has been thought to have no anti-HIV activity. Recent case reports suggest ETV may have some activity against HIV, but further studies are needed to confirm these observations.
Mutations in HBV polymerase that lead to 3TC resistance also cause ETV resistance and higher doses of ETV are required (0.5 mg daily for 3TC-naïve and 1.0 mg daily for 3TC-resistant patients) (Soriano, 2005).
Another nucleotide analogue of adenosine monophosphate, tenofovir disoproxil fumarate (TDF), is licensed for the treatment of HIV but also has activity against both wild-type and 3TC-resistant HBV. In a study of both 3TC-resistant and 3TC-naïve HBV-HIV coinfected subjects, Peters and colleagues showed that daily 300-mg dose of TDF was as effective as 10 mg of ADV (Peters, 2006). Treatment with either drug resulted in clinically important HBV DNA suppression and improvement in ALT without loss of HIV control.
Telbivudine (LdT) is a recently-approved nucleoside analogue of thymidine with anti-HBV activity and no anti-HIV activity. It causes a more profound reduction in HBV DNA than 3TC (Lai, 2005); however, YMDD mutations of the HBV polymerase gene also cause LdT resistance (Soriano, 2005), limiting its use in 3TC experienced patients.
Several studies have shown that combination therapy is effective for the treatment of HBV in HBV-HIV coinfection. Benhamou and colleagues showed that ADV when added to a HAART regimen including 3TC resulted in significant reductions in HBV DNA (P<.0001) and ALT (P<.05), and these effects increased with treatment duration (Benhamou, 2006). Despite 3TC-resistant HBV in greater than 90% of the subjects in the TDF/ADV non-inferiority study described above, 96% were taking 3TC as part of their HAART regimen during the study. Therefore, this study also examined the effects of combination therapy in HBV-HIV coinfection (Peters, 2006).
Therapy for HBV should be considered in HBV-HIV coinfected patients with evidence of liver disease (elevated ALT, elevated HBV DNA, or necroinflammation and fibrosis in liver biopsy) (Soriano, 2005). The choice of HBV therapy for patients with HBV-HIV coinfection should be based on the need for concomitant HIV therapy (Table 6) (Peters, 2006).
| Table 6. Therapy and Monitoring of HBV-HIV Coinfection. | |
| Therapy | Therapy |
| Choose drug depending upon need for hiv therapy | alt every 3 months |
| Long-term therapy is the rule | hbv dna every 3 months |
| Combination therapy recommended | Increase in hbv dna If on therapy: drug resistance and/or viral breakthrough? If not on therapy: reactivation? |
| Always add anti-hbv drug to haart | hcc screening every 6 months: afp + imaging |
| afp, alpha fetoprotein; alt, alanine aminotransferase; haart, highly active antiretroviral therapy; hbv, hepatitis B virus; hcc, hepatocellular carcinoma; hiv, human immunodeficiency virus | |
When treatment of HIV is not indicated, pegylated interferon should be considered because it does not lead to drug resistance in HIV or HBV. Interferon is more effective in the treatment of HBeAg-positive than -negative patients. Treatment response rate is poorer in HBV-HIV coinfected patients compared with HBV monoinfected patients. It should be used with caution in patients with compensated cirrhosis and is contraindicated in patients with decompensated cirrhosis (Soriano, 2005). Because ETV and ADV are not active against HIV, they are also recommended for the treatment of chronic HBV when the treatment of HIV coinfection is not indicated (DHHS, 2006; Peters, 2006; Soriano, 2005). However, the potential for the development of HIV drug resistance while on ETV monotherapy for chronic hepatitis B was recently reported in a patient with a prior history of HIV treatment with HAART (FDA, 2007).
Telbivudine can also be used, although it has not yet been incorporated in published treatment guidelines. The use of drugs with activity against both HIV and HBV (3TC, FTC, or TDF) should be avoided without HAART because of rapid development of drug-resistant HIV strains (DHHS, 2006; Peters, 2006).
When treatment of HIV is indicated, patients with HBV-HIV coinfection should always receive anti-HBV agent with initiation of HAART to avoid the risk of decompensation due to immune reconstitution. For coinfected patients, combination therapy with a nucleoside analogue (3TC or FTC) and TDF is recommended as part of the HAART regimen (DHHS, 2006; Soriano, 2005). To avoid development of HBV resistance, 3TC, or FTC should not be used as the only active anti-HBV agent (DHHS, 2006).
| Monitoring | Top of page |
All HBV-HIV coinfected patients should be monitored with serum ALT every 3 months (Table 6). Those undergoing therapy need to be monitored for viral breakthrough and development of drug resistance. Because HBV is a dynamic disease, patients who do not require treatment still require monitoring every 3 months to recognize reactivation and subsequent need for treatment. As previously discussed, this includes patients who have seroconverted (who have developed anti-HBe or anti-HBs) because they remain at risk for reactivation. Patients with HBV-HIV coinfection should be screened for HCC every 6 months with serum alpha fetoprotein and ultrasonagraphy or 4-phase abdominal computed tomography (Brook, 2005; Soriano, 2005).
In HBV monoinfection, an early antiviral response at 12 to 24 weeks is strongly linked to subsequent outcomes on therapy. Patients with undetectable HBV DNA at week 24 are very likely to remain undetectable at week 52, while patients with greater than 3 log10 copies per mL of HBV DNA at week 24 have a high likelihood of viral breakthrough and drug resistant mutations at week 52 (Lai, 2005). Similarly, the rate of HBeAg loss is related to the level of HBV DNA at week 24, with over 90% of patients with greater than 3 log10 copies per mL of HBV DNA failing to seroconvert by week 52 (Lai, 2005). Therefore, if there is a limited response to therapy after 12 to 24 weeks, a change should be made by adding or replacing a drug.
| Treatment Failure Due To Antiviral Resistance | Top of page |
Figure 3.Lamivudine Resistance in HBV Monoinfection.
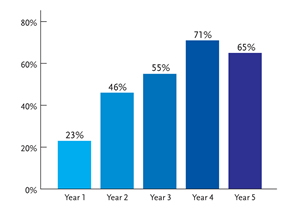
Proportion of rtM204V/I mutations in lamivudine-treated patients. Reprinted with permission from Lok AS, et al. Gastroenterol. 2003;348:808-816.
The main cause of anti-HBV treatment failure is the selection of resistant HBV variants. Poor patient compliance to treatment and persistence of covalently closed circular DNA (cccDNA, the template for pregenomic RNA) are also important causes of treatment failure, but will not be discussed here.
The initial emergence of 3TC-resistant HBV mutations developed from the frequent use of 3TC in most HAART regimens for HBV-HIV coinfected patients. The most important mutations are rtL180M and rtM204V/I, with the latter occurring in the YMDD motif of the reverse transcriptase domain of HBV polymerase. The cumulative incidence of 3TC resistance in HBV-monoinfected patients increases with duration of treatment, from 23% in the first year to 71% after 4 years (Figure 3) (Lok, 2003).
Most 3TC-resistant variants have reduced fitness compared with wild-type HBV leading to lower HBV DNA levels compared with pretreatment (Lai, 2003; Lok, 2003; Pillay, 2000). However 88% of individuals with 3TC-resistant mutations also have compensatory mutations leading to similar HBV DNA levels as noted pre therapy (Delaney, 2003). In a placebo-controlled trial of 3TC therapy, Liaw and colleagues demonstrated a marked reduction in disease progression and the risk of liver complications with 3TC compared with placebo (Liaw, 2004). These benefits of 3TC were observed despite the development of 3TC-resistant mutations in nearly half the subjects assigned to the 3TC arm. However, subjects with 3TC-resistant mutations were more likely to have an increase in the Child-Pugh score and die from a liver-related death than subjects without 3TC-resistant mutations. It was recognized that although HBV DNA levels did not reach pretreatment levels, the emergence of 3TC resistance led to resumption of viral replication and disease progression.
Initially, 3TC was the only approved oral therapy for HBV, and interferon was contraindicated for the treatment of patients with advanced fibrosis or cirrhosis due to the risk of hepatic decompensation. Despite the high incidence of 3TC resistance, it was better than placebo (Liaw, 2004), and therapy was often continued despite the emergence of resistance.
In a retrospective analysis of clinical trial data involving treatment with 3TC for up to 6 years, Lok and colleagues found that 3TC resistance was associated with an increase in hepatitis flares (Lok, 2003). The frequency of hepatic decompensation and liver-related death in patients with 3TC resistance was similar to patients without resistance, but this appeared to increase with time. These findings suggest that, over time, the initial clinical benefit of treatment with 3TC is lost in patients who develop 3TC resistance.
When ADV was introduced, it was touted for its efficacy against 3TC-resistant HBV (Perrillo, 2000) and for the absence of resistance in 2 large 48-week trials (Hadziyannis, 2003; Marcellin, 2003). However, with increased use of ADV, resistant viruses emerged (eg, rtN236T/rtA181V) (Angus, 2003). In one study, even though the incidence of ADV resistance was 0% after one year, the cumulative incidence after 5 years was 29% (Hadziyannis, 2006). Higher HBV DNA at week 48 during ADV therapy predicted the emergence of ADV resistance, as has been noted with 3TC and LdT.
Antiviral drug resistance reflects reduced susceptibility of a virus to the inhibitory effect of a drug and results from a process of adaptive mutations under therapy. Factors that affect the emergence of antiviral resistance are drug selective pressure, the fitness of the variant, and the turnover of host cells (Pillay, 2000). Hepatitis B-infected hepatocytes have a half-life of 10 to 100 days compared with 2 days for HIV-infected CD4 cells, which might explain the relatively slow process of antiviral resistance seen in HBV compared with HIV. The emergence of resistant mutants in HBV, therefore, is accelerated by high HBV replication, and hepatocyte necroinflammation and proliferation (Zoulim, 2001). Viral kinetic studies in HBV mono-infected patients with 3TC resistance have confirmed the association of high baseline HBV DNA and ALT levels with the emergence of 3TC resistance (Lai, 2003; Si Ahmed, 2000; Yuen, 2001). Genotypic resistance precedes an increase in HBV DNA (phenotypic resistance) by about 3 to 6 months, and patients with HBV DNA levels greater than 103 copies per mL at 6 months of therapy are more likely to develop YMDD mutations leading to HBV DNA breakthrough. Other predictors of the emergence of YMDD variants and the development of 3TC resistance include treatment duration, high body mass index, and male sex (Lai, 2003).
The emergence of antiviral resistance is indicated by increasing HBV DNA, defined as rebound of HBV DNA of at least 1 log10 IU per mL* from nadir in patients with an initial antiviral treatment effect (Locarnini, 2004). Increasing ALT levels also indicate the development of resistance, though this is usually a late sign. Aminotransferase elevations associated with antiviral resistance in HBV are preceded by elevations in HBV DNA due to the host immune response, which is in contrast to HIV where the emergence of antiviral resistance is associated with an increase in HIV viral load and concomitant decrease in CD4 cell count. It is now known that continued viral replication in the face of antiviral resistance leads to an increase in flares and progression of liver disease over time (Lok, 2003), a situation exacerbated when therapy is continued after the development of resistance. This underscores the importance of changing therapy when there is limited or no response.
| Compensatory And Vaccine Escape Mutations | Top of page |
The in vitro characterization of viral mutations has demonstrated the emergence of compensatory mutations that enhance the reduced viral replication of primary resistance mutations. For example, the mutation rtV173L, located near the HBV polymerase active site, increases the replication efficiency of 3TC-resistant HBV (rtL180M, rtM204V) to near wild-type levels (Delaney, 2003). Compensatory mutations in regions distant from the HBV polymerase active site can result in conformational changes that can affect the cross-sensitivity profile of several antiviral agents and confer potential multidrug resistance (Bartholomeusz, 2005).
Due to the overlapping reading frames of the genes encoding HBV polymerase and HBsAg, mutations in one gene may affect the other. If these mutations affect the neutralization domain (the “a” determinant) of the HBsAg, HBV vaccine (anti-HBs) efficacy will be compromised. In a cohort of 3TC-treated HBV-HIV coinfected patients, the triple polymerase mutant (rtL173V, rtL180M, rtM204V) was found in 17% of patients (Matthews, 2006). The frequency of this mutation increases with duration of 3TC treatment. This and other mutations, known as vaccine escape mutations, create antigenically altered HBsAg proteins and demonstrate markedly reduced binding to anti-HBs (Torresi, 2002). These mutations may also affect the ability of current assays to detect HBsAg and compromise the ability to diagnose HBV (so-called diagnostic escape mutants). This might be one underlying explanation of occult HBV infection.
| Conclusion | Top of page |
HIV substantially impacts the outcome of HBV. Although ALT levels are lower in HBV-HIV coinfection, liver damage quietly, yet rapidly, progresses. With improved control of HIV disease with HAART, liver disease has emerged as one of the leading causes of death in patients with HIV.
HBV infection is a dynamic disease and coinfection with HIV considerably complicates its diagnosis and management. The choice of antiviral therapy should be based on the need for HIV therapy, with control of HBV when HAART is initiated. Combination therapy should be used to avoid development of antiviral resistance. Continuous monitoring of HBV patients, regardless of need for treatment or history of seroconversion, is imperative to recognize reactivation and subsequent need for treatment, and to identify drug resistance and viral breakthrough early. Prompt changes in therapy when resistance emerges will reduce the development of compensatory mutations that will affect our ability to use newer therapies and lead to transmission of drug-resistant viruses in vaccinated individuals.
In many respects, the treatment of HBV is a step behind HIV which has embraced combination therapy with a variety of drug targets. Unfortunately only nucleos(t)ide analogs are as yet available for HBV but much has been learned from the lessons of HIV treatment. The complexities of HBV-HIV coinfection highlight the importance of close working relationships between hepatologists, infectious disease specialists and primary-care providers in order to optimize patient outcomes.
| References | Top of page |