
Patients who have developed resistance to the 3 currently available classes of HIV medications present a particular treatment challenge. Developments in an entirely new class of HIV antiretroviral therapy, inhibitors of the viral integrase protein, were the focus of discussions at a recent PRN meeting.
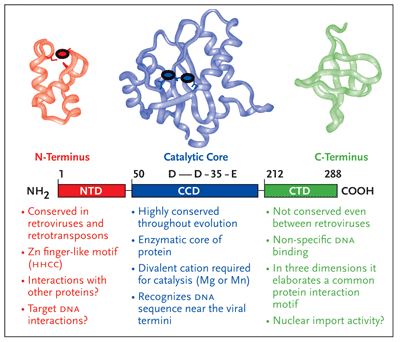
HIV-1 integrase is a viral protein that has several roles (Figure 1). After the completion of reverse transcription of the viral RNA genome into DNA, it appears in part, to be responsible for ferrying the viral DNA into the nucleus of the cell, a position where it has the ability to closely associate with one of the chromosomes of the host cell. Subsequently, integrase catalyzes the insertion, or integration, of viral DNA into the host cellular DNA, a step of the viral lifecycle required for reprogramming the cell to produce more copies of itself (Goff, 1992; Kulkosky, 1994; Lewinski, 2005). Integration is, in effect, the most intimate association any virus can make with its host the irreversible chemical fusion of its DNA with that of the cell. Chromosomal DNA then serves as a sanctuary for the expression of viral information as well as the perpetuation of the retroviral genome. Once integrated into the host chromosome, retroviral DNA is a permanent fixture and upon cell division, is passively transmitted as an embedded passenger within the DNA of all daughter cells. Thus, the infected cell is a predisposed source of virus production not only for the lifetime of that cell but for all subsequent generational derivatives (reviewed in Coffin, 1997).
HIV-1 integrase is a member of a protein superfamily known as the polynucleotidyl transferases (Mizuuchi,1997) (also see sidebar on page 10). It has an ancient and highly conserved catalytic domain, described not only in human retroviruses but also found in the retroviral-like elements of the fruit fly, Drosophila and yeast (Flavell, 1997), and even exists in certain primitive transposons found in bacteria (Hallet and Sherratt, 1997; Polard, 1995; Rice,1996). Perhaps paradoxically, the formation of the human immune system also requires a similar functional domain as a part of the RAG recombinase system (Jones, 2004; Schatz, 2004). The RAG enzymes are responsible for the maturation of B and T cells, which in turn, are critical to fight against invading microorganisms, like HIV-1.
In the context of the HIV lifecycle, the integration event itself divides the viral life cycle into 2 distinct phases (preintegration and postintegration)
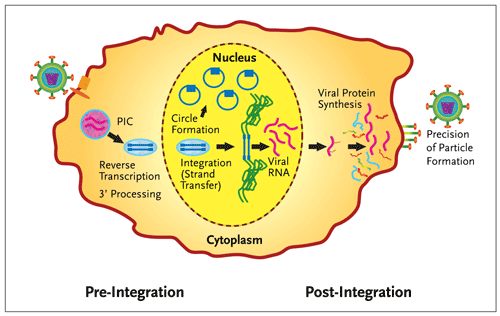
Figure 1.Integrase Divides the Retroviral Lifecycle.
Integration of HIV-1 DNA (blue) into the host chromosome (green) divides the retroviral life cycle. In addition to the catalytic functions required for integration (3 processing and strand transfer), integrase is also involved in other essential functions (reverse transcription, the circularization of the viral DNA into extra-chromosomal forms and the precision of virion particle formation). PIC is the preintegration complex, an operationally defined association of viral and host proteins that coordinate reverse transcription and nuclear import of the viral genome. In this figure, viral entry is on the left and egress of newly formed particles on the right.
However, there are other steps either preceding or immediately after the step of chemical bonding between the viral and host DNAs that are equally essential for the completion of this complicated set of enzymatic reactions. First, while still in the cytoplasm, integrase cleaves 2 nucleotides from the ends of newly reverse transcribed and linear viral DNA. This reaction (the 3' processing reaction) generates a reactive chemical group (3' hydroxyl) at each terminus of the viral DNA (Figure 2).
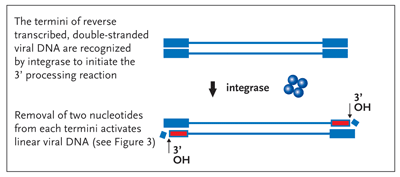
Figure 2. The 3Processing Reaction Activation of the Viral DNA Termini
Following the completion of reverse transcription of the viral RNA into its DNA copy, integrase (here shown as blue tetramer) removes two nucleotides from the 3 end of each strand of the viral DNA generating a chemically reactive hydroxyl group (3OH, red). This reaction serves to activate the termini of the linear dna molecule for a subsequent strand-transfer step outlined in Figure 3, the target of all integrase inhibitors currently in the clinic.
Integrase is then imported into the cell nucleus by an unknown mechanism within the context of a preintegration complex, or PIC. The PIC is composed of viral and host proteins in conjunction with the viral DNA. The nuclear (viral) DNA then undergoes one of two fates. In the strand-transfer reaction (Figure 3), the activated 3' hydroxyl groups left after the 3' processing reaction attack the host chromosome on opposite sides of the DNA helix, producing a 5-base-pair staggered cut, simultaneously forming stable chemical bonds between the inserted viral DNA and that of host cell DNA. Finally, the ragged ends produced on opposing sides of the sealed recombinant joint are repaired by as yet unknown host factors and the integrity of the chromosomal DNA restored.

Figure 3. The Strand Transfer Reaction Intimate and Irreversible Bonding to Host Cell DNA
Integrase utilizes the reactive chemical groups produced in Figure 2 (red, 3 OH) to simultaneously make a 5-base-pair staggered cut in the host chromosomal DNA (green) and then to stably introduce the viral DNA at the cleaved sites. This occurs as a concerted reaction and is reversible in vitro. However, once the integrity of the chromosomal DNA is restored by the repair of the recombinant joint by the action of undefined host factors (man-at-work sign), the viral DNA insertion becomes indistinguishable from the rest of the cellular dna and cannot be removed.
It is amazing that the integrated viral DNA copy, comprising only about 0.0003% of the entire genetic capacity of the cell, can now commandeer the cellular environment to its own advantage. The other fate of nuclear viral DNA is the circularization of these linear molecules into extrachromosomal forms (Figure 4). They are the products of the activities of cellular enzymes in the nucleus. Although the function of the circles with regard to their contribution to the viral lifecycle is not yet understood, it has been known for some time that these molecules can act as templates for limited expression of the viral genome (although clearly not enough to produce all of the viral proteins necessary to assemble the mature virus) (Wiskerchen, 1995; Wu, 2001; Wu, 2003). Whether or not the low level of viral gene expression originating from the circles contributes to the pathogenesis of the virus remains to be determined.
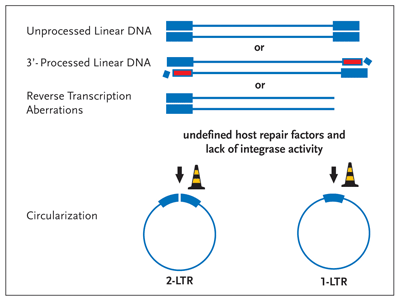
Figure 4. Circularization— An Alternative Fate of Linear Viral DNA
Linear viral DNA is circularized by host nuclear proteins in the absence of integrase enzymatic activity. If integrase fails to process the 3 termini (Figure 2) or if the enzyme fails to perform the strand transfer reaction (Figure 3), the viral DNA is circularized by the activity of still ill-defined host proteins (road work cones). Although these circular DNA molecules can act to produce a subset of viral proteins, they can't provide for the synthesis of the viral structural gene products, key to virion formation and infectivity
| Therapeutic Discoveries | Top of page |
In light of the inability of HIV to propagate itself without integration of its genome into chromosomal DNA, the viral integrase protein has become an important potential therapeutic target. It is distinct mechanistically, independent of the catalytic activities of the viral protease, reverse transcriptase, or of virus cell fusion. It therefore has the potential for synergistic activity in combination with available protease and reverse transcriptase inhibitors, and with entry inhibitors now in development. In addition, because of their novel molecular structure, the cost of production of the new families of integrase inhibitors may be less than that of some of the current antiretrovirals which rely on protein-like compounds or nucleoside derivatives for their activity. Although it has been more than a decade since integrase was first described, it has been very difficult to find therapeutic inhibitors. For instance, the integrase protein is insoluble in its native form and it has been relatively difficult to purify and study its biochemical and physical properties. It is anticipated that, given the properties of the enzyme, it may well be that several forms of the protein may exist, perhaps with each structural configuration relevant to a specific activity of the protein. The search for new inhibitors has also been confounded by the fact that many of the multiple compounds that have been described as inhibiting the activities of integrase in vitro fail to exhibit specific activity against integration during viral infection in vivo. Indeed, very few of the lead compounds found in vitro can satisfy the 4 critical qualifications that were recently outlined (Pommier, 2005). These criteria are: 1) time of drug addition experiments that must show that the drug is active after the completion of reverse transcription (about 4-6 hours postinfection [hpi]) but not after 10 to 12 hpi, the approximate timing of HIV-1 integration; 2) there must be a concomitant rise in the accumulation of circular species in treated cells indicating that the drug specifically inhibits HIV integration allowing for the alternative fate of viral DNA (circularization) to predominate; 3) the location of mutations associated with drug resistance must map to integrase; and 4) purified mutant integrase protein must exhibit associated drug resistance in standard biochemical assays for the strand transfer reaction. Of great assistance to the development of potential compounds was the assay system to detect the specific inhibition of the integrase-catalyzed strand transfer reaction (Hazuda, 2000). Among the inhibitors screened for activity, a set was found that led to the specific inhibition of HIV integration (the strand transfer reaction) and interrupted replication in tissue culture cells.
Subsequently, Dr. Hazuda and colleagues published the result of a proof-of-principle trial that demonstrated that compound L-870,810, a naphthyridine carboxamide could inhibit retroviral replication in rhesus macaques (Hazuda, 2004). Other groups have also developed potential drugs, most of which are thought to act by inhibiting strand transfer. No compounds that inhibit the 3' hydroxyl processing reaction are in human clinical trials. Merck-0518 and Gilead-9137 are currently being evaluated in the clinic. They are active in the nanomolar range, their activity generated, in part, from bond formation between metal ions contained at the active site of the enzyme and the compound (Grobler, 2002). Mutations originating from drug resistance studies of similar integrase inhibitors cluster near a region in the 3-dimensional structure near the active site and overlapping a region of residues that appear to bind DNA. Interestingly, there may be a requirement for drug binding in the context of a ternary complex, which means that the drug might interface not only with integrase, but also with the cations and nucleic acids that are present at the active site.
| Findings From Clinical Trials | Top of page |
Although integrase inhibitors have been in development for a long time, there have been many hurdles to discovering effective, nontoxic compounds. The new compounds currently under investigation have shown great activity both in vitro and in clinical trials. Their arrival into the panoply of antiretroviral medications should have a similar impact to that of protease inhibitors 10 years ago. There are currently 2 integrase inhibitors undergoing phase 2 or later clinical trials. These compounds are described below.
Merck-0518
Merck-0518 is a new compound, a naphthyridine carboxylase with potent in vitro activity in the nanomolar range. It is metabolized by glucuronidation (UGT1A1), and does not have any significant effect on CYP3A4; it therefore has limited interactions with most medications. Furthermore, it has demonstrated good in vitro synergy with available antiretrovirals, and there is no evidence from in vitro studies of crossresistance with other medications (Markowitz, 2006). Merck-0518 has an excellent safety profile in preclinical studies which has been confirmed at different doses in normal volunteers.
In a small, randomized, double-blind, placebo-controlled study of Merck-0518 versus placebo, 35 patients who were HIV infected and antiretroviral (ART) naive were given monotherapy for 10 days (Markowitz, 2006). The endpoints were virologic response and adverse events. At entry, patients had viral loads greater than 5,000 and CD4 cell counts greater than 100 cells per mm3. Exclusion criteria were any underlying liver, renal, or immunosuppressive disease. The pharmacokinetics during the 10-day monotherapy study demonstrated little difference in the areas under the curve (AUCs), the trough levels, or in the maximum concentration (Cmax) of the different doses. At all doses, there was an approximately 2-log reduction in viral load by 10 days, and by the end of the study, about a quarter of the patients had undetectable viral loads (Figure 5). During this period, there was little change in the CD4 cell count.
Adverse events, which included dizziness, fatigue, and headache, were generally mild and transient, and none were dose related. These adverse events were also present in the placebo group.
The trial was then extended as a phase 2b trial and those enrolled in the monotherapy group were given the option of continuing. During this phase, patients were randomized to triple therapy with a tenofovir and lamivudine backbone, and either efavirenz or 1 of 4 different doses of Merck-0518. Based on pharmacokinetics and antiretroviral activity, the optimal dosing has been shown to be 400 mg twice daily. This study is ongoing.
A parallel study in patients who have documented triple class antiretroviral resistance has also been initiated. In this study, different twice-daily doses of Merck-0518 were compared with a placebo, all in combination with an optimized background. Patients were stratified on the basis of T20 use in the optimized background, and on the baseline degree of HIV resistance to protease inhibitors.
As shown in Table 1, there were about 40 patients in each arm, predominantly male, with a median age of approximately 44 across all arms, as one would expect in a highly treatment-experienced population. Baseline viral loads were approximately 50,000 and the mean CD4 cell count was 220 to 283. The median number of years of antiretroviral therapy was 9 to 11, again signifying that this was a drug-experienced population with a lot of antiretroviral exposure.
| Table 1. Baseline Patient Characteristics | ||||
| 200 mg N=40 |
Merck-0518* 400 mg N=42 |
600 mg N=42 |
Placebo* N=43 |
|
| Median Age (yrs) | 43 | 44 | 44 | 43 |
| Male | 83% | 91% | 91% | 88% |
| Mean log10 HIV RNA | 4.6 | 4.8 | 4.7 | 4.7 |
| Mean cd4 Count (/mm3) | 244 | 220 | 226 | 283 |
| Median Years of Prior ARTs | 9 | 11 | 9 | 10 |
| OBT: Median # of ARTs | 4 | 4 | 4 | 4 |
| OBT: # of pts using T-20 (%) | 13 (33%) | 16 (38%) | 16 (38%) | 16 (38%) |
| PSS†: 0 to all ARTs | 16 (40%) | 24 (57%) | 21 (50%) | 17 (40%) |
| PSS†: 0 to PI | 39 (98%) | 40 (95%) | 37 (88%) | 36 (84%) |
| * + OBT; † PSS = Phenotypic sensitivity score; Enfurvitide is not included in the scores since there is no clinical cut-off. Reprinted with permission from Grinsztejn B et al. 13th Conference on Retroviruses and Opportunistic Infections. February 5-8, 2006, Denver, Colorado. Abstract 159LB. |
||||
This is reflected by the very low phenotypic sensitivity score (PSS) across all arms; 40% to 60% of patients had a PSS of zero to all arts, not including T20, indicating that they were not sensitive to any of the antiretrovirals used in the background regimen. Furthermore, only about a third of patients used T20 as part of their background therapy. This implies that, in many of the patients, the activity of the integrase inhibitor is being seen alone, although only over a relatively short period.
By week 16, the discontinuations were very few: 1 patient due to lack of efficacy and 1 due to suicide. There was also a substudy done to evaluate a potential interaction with atazanavir, a UGT1A1 inhibitor. The treatment effect across arms A and B were identical; therefore, the data have been pooled (Grinsztejn, 2006).
The viral impact of Merck-0518 was seen quickly: many patients had achieved a viral load of less than 400 copies per mL by week 4 of therapy. By week 8, 63% to 67% of patients receiving Merck-0518, versus 8% receiving placebo with their optimized background therapy (OBT), had achieved a viral load of less than 50 copies per mL. This effect persisted until week 16, where 50% to 60% of patients maintained an undetectable viral load at all 3 doses (Figure 5).
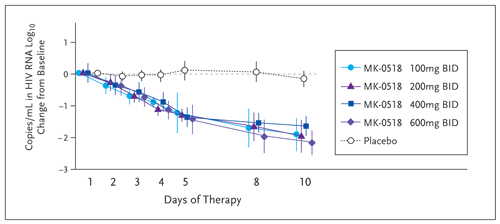
Figure 5. Change from Baseline in HIV-1 RNA Following Monotherapy with Merck-051
Reprinted with permission from Markowitz M, et al. 16th International AIDS Society Conference. April 13-18, 2006. Toronto, Canada. Abstract THLB0214
In comparison, the OBT with placebo had an average 1-log reduction in viral load, consistent with other studies of OBT. The increase in CD4 T-cell count was modest, but comparatively good when the population under study is considered (Grinsztejn, 2006).
Similarly to the short-course monotherapy study, there were very few adverse events attributable to the study drug. In terms of short-term tolerability, there was little difference between the Merck-0518 groups and placebo. Most of the clinical adverse experiences were mild to moderate, with few discontinuations by week 16.
With regard to laboratory abnormalities, there was some increase in triglycerides and cholesterol, but this result was confounded by the presence of boosted protease inhibitors in the OBT. There were grade 3 to 4 elevations in total bilirubin, but this was only seen in patients also taking atazanavir. Encouragingly, there was no evidence of dose-related toxicity in the integrase inhibitor arms in this challenging patient population. Merck-0518 has demonstrated potent antiviral activity with up to 72% of patients with a viral load of less than 50 copies per mL at week 16, and limited toxicity.
In light of the excellent results of these 2 studies, Merck is continuing the multicenter phase 2b trial comparing Merck-0518 versus efavirenz, and is also doing an international phase 3 study in patients with multidrug-resistant viruses. There are approximately 650 patients enrolled in clinical trials evaluating the efficacy of 400 mg bid of Merck-0518 versus placebo, in conjunction with an optimized background regimen. Patients are able to take enfuvirtide, tipranavir and/or darunavir as part of their regimen, allowing a thorough assessment of this compound as part of current salvage therapy. Merck has recently made Merck-0518 available under an expanded access program.
Gilead-9137
Gilead-9137, a dihydroquinoline carboxylic acid originally developed as Japan Tobacco 303, is now in phase 2 clinical trials. It is also a strand transfer inhibitor thought to act by interacting with the divalent metal ions in the catalytic core domain. It has potent activity in vitro, with a serum inhibitory concentration of 50% (IC50) of 0.2 nM against HIV-1, and a protein-binding adjusted IC50 of 16 nM. Like Merck-0518, Gilead-9137 is also active against multi-drug resistant virus. Animal studies demonstrated a maximum dose of 2,000 mg per kg per day, with no evidence of dose-limiting toxicities.
According to preclinical studies of pharmacokinetics, Gilead-9137 undergoes both oxidative metabolism via CYP3A and glucuronidation. It is an inducer of CYP3A, and therefore has the potential for interactions with other compounds. In studies of unboosted Gilead-9137, it has a relatively short half-life of 3 hours, and a higher systemic exposure (AUC) when dosed with food. The pharmacokinetics support bid dosing (DeJesus, 2006). However, there is a dramatic increase, one approximately 20-fold higher, in the AUC when 100 mg of ritonavir is added. The plasma half-life also increases to 9 hours, allowing once-daily dosing. Furthermore, there is no autoinduction of metabolism with ritonavir.
Results of a randomized, placebo-controlled monotherapy study of 40 patients who were HIV-1 infected and treatment naive or experienced were recently published (DeJesus, 2006). Patients who were not currently taking ART and who had a viral load between 10,000 and 30,000 copies per mL with a CD4 cell count above 200 cell per mm3 were randomized to 1 of 5 dosing groups or placebo. These criteria were in place to minimize the rapid selection of resistant virus in people with advanced disease. The dosing groups included 200, 400, or 800 mg bid, or 800 mg qd, or 50 mg qd boosted with 100 mg of ritonavir. Patients received 10 days of therapy getting either compound or placebo. Pharmacokinetics and viral response were evaluated on days 1 through 10, and the patients were followed for 11 days off treatment with serial trough sampling (Figure 6). The endpoints of the study were changes in HIV RNA, safety and tolerability, and pharmacokinetics. All Gilead-9137 dosing regimens were well tolerated. Any adverse events were grade 1 or 2 in severity, were not dose related, and resolved on treatment. Clinical adverse events included fatigue, diarrhea, and headache, but were also present in the placebo arm. There were no study drug discontinuations. The pharmacokinetic studies demonstrated that the addition of 100 mg of ritonavir to 50 mg of Gilead-9137 boosted the peak level to one roughly equal to the twice-daily dosing, with a far superior trough.
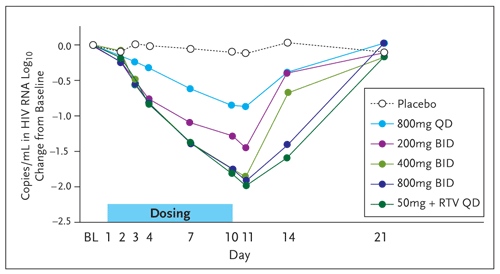
Figure 6. Change from Baseline in HIV-1 RNA Following Gilead-9137 Monotherapy
Reprinted with permission from DeJesus E, et al. J Acquir Immune Defic Syndr. 2006;43:1-5.
Importantly, the addition of ritonavir allowed a more potent antiviral effect with much less drug. Although there was a very good reduction in viral loads across all dosing groups, the 50-mg daily dose with ritonavir achieved a 1.5 to 2.4 log10 drop in HIV-1 RNA. This led to the decision to further develop Gilead-9137 with ritonavir boosting. Gilead is currently conducting a phase 2b trial comparing Gilead-9137 at different doses (20, 50, or 125 mg daily with ritonavir) with an OBT versus a boosted protease inhibitor with an OBT. This is being done in patients who are treatment experienced and who have documented resistance to protease inhibitors.
Resistance to Integrase Inhibitors
To date, there is little known about in vivo resistance to integrase inhibitors. In vitro data show varying patterns of resistance between different classes of compounds. Resistant viruses very slowly emerge in vitro. However, single amino acid substitutions may be associated with substantial resistance. Encouragingly, these single-point mutations appear to make the viruses much less fit. Resistance testing that was done at baseline and during viral rebound post-treatment in Gilead-9137 trials revealed no integrase inhibitor resistance mutations at either point.
Future Directions
As the clinical development of integrase inhibitors progresses rapidly, one must ask where these new agents will fit in the treatment armamentarium. Preferred treatment for NNRTI-susceptible, treatment naive patients is likely to remain one pill-once daily. However in some patients such as the NNRTI resistant or intolerant, integrase inhibitor therapy may become an option if apparent lack of toxicity of Merck-0518 is sustained. A note of caution however, though Gilead-9137 is being developed as a once daily drug the risk of using ritonavir-boosting in the absence of a protease inhibitor containing regimen remains unknown. Like most new agents, integrase inhibitors will initially be used in the setting of treatment failure. With the availability of novel protease inhibitors as well as the prospect of more orally bioavailable entry inhibitors the prospect of creating salvage regimens with 3 new agents is rapidly becoming a reality- and the goal of sustained suppression of viral replication in this challenging patient population may be achievable in the majority of treated patients. Finally, it will be very interesting to see how resistance to integrase inhibitors emerges in the context of preexisiting multidrug-resistant virus. Hopefully, the virus will have a limited capacity to compensate for developing resistance to multiple classes of antiretrovirals, and that any resulting virus will be weak, unfit, and difficult to transmit.
| References | Top of page |

{kind=link}
{kind=link}