
Recent studies in HIV-1 infected individuals have demonstrated that the gastrointestinal (GI) tract is preferentially and profoundly affected during acute and chronic HIV-1 infection. These findings shed light on early postinfection events at mucosal sites. In addition, they raise important questions regarding the long-term impact of immune depletion within the GI tract on HIV-1 pathogenesis. The following review summarizes recent studies that have brought the GI immune system to the forefront of HIV research.
| The Gastrointestinal Tract as an Immunologic Organ | Top of page |
The primary function of the GI tract is digestion, absorption, and assimilation of nutrients (Table 1). To maximize the efficiency of digestive function, the GI tract has evolved such that it has the largest surface area among all organs. In fact, the 400 m2 surface area of the GI tract is about 200 times larger than the surface area of the entire skin.1 Having such a large surface in close proximity to the external environment necessitates that a large complement of immune cells—both innate and adaptive—be deployed at mucosal surfaces. Indeed, GI-associated lymphoid tissue constitutes the largest immune compartment in the body. It is estimated that T cells associated with the small intestinal epithelium alone may account for more than 60% of the total body lymphocytes.2
| Table 1. Salient Features of the Gastrointestinal Immune System |
| The largest reservoir of immune cells in the body. |
Organized into:
|
Antigen is presented via various pathways to inductive sites in
the GI tract, including the uptake of antigen by specialized “M”
cells.
|
Immune cells in the GI tract are organized into distinct anatomic and functional subcompartments (Figure 1).3 These are classified as immune inductive sites, considered as sites of T cell education, and immune effector sites, where T cells neutralize foreign antigens—both microbial and nonmicrobial. Immune inductive sites are comprised of Peyer’s patches (PPs) and mesenteric lymph nodes (MLNs). Peyer’s patches have the anatomic appearance of secondary lymphoid organs, with clearly defined T- and B-cell–dependent areas. A single layer of epithelial cells separates the PPs from the intestinal lumen. This epithelial cell layer contains specialized cells called M cells in addition to conventional enterocytes. It appears that the function of M cells is transport of antigen into immune inductive sites resulting in initiation of T-cell education and maturation. It is believed that further maturation of T cells continues within MLNs that drain GI mucosal tissue. It is thought that MLNs are the crossroads of systemic and mucosal immunity. Within the MLNs, lymphoid cells from mucosal and systemic immunity interact, T-cell maturation continues, and it is likely that critical issues of gut homeostasis are determined as outlined below.
Immune effector sites may be thought of as the battlegrounds where terminally differentiated T cells perform effector function, neutralizing antigen and protecting the host against invading pathogens. Effector site lymphocytes can be subclassified into lamina propria lymphocytes (LPLs) and intraepithelial lymphocytes (IELs). As detailed below, LPLs have a unique phenotype—these cells are terminally differentiated effector T cells that have pathways of stimulation that are different from peripheral blood T cells. For example, it appears that LPLs proliferate at relatively low levels in response to antigen or other T-cell antigen receptor (TcR)–dependent stimuli.4,5 In contrast, LPLs secrete large amounts of effector cytokines, such as IFN-γ, IL-4, and IL-5 4,6, and have a unique requirement for CD2-dependent pathways of activation 7. The IELs also constitute a unique population of lymphocytes in the body. More than 80% of IELs are CD8+ and a substantial proportion of IELs express the CD8 αα homodimer in contrast to the CD8 αβ heterodimer expressed on a majority of peripheral blood CD8+ lymphocytes.2,8,9
Figure 1. Organization of the Intestinal Immune System.
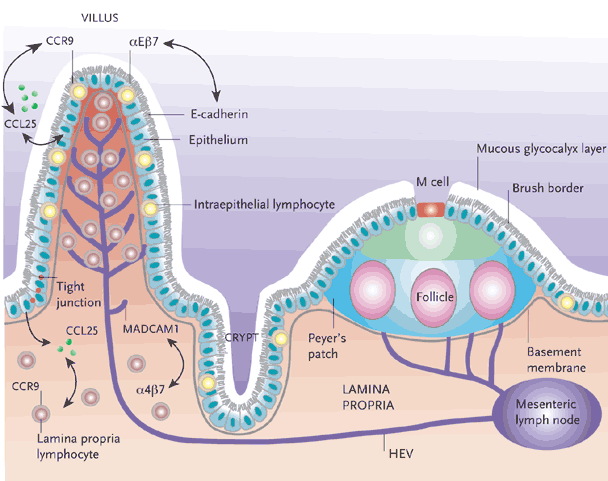
The effector compartment consists of lamina-propria lymphocytes and the IELs. M cells act as specific entry ports for antigen, facilitating the uptake of luminal antigens and their subsequent delivery to the initiation compartment of the organized lymphoid tissues, including the pps and the MLNs. Naive T cells activated in the PPs and MLNs upregulate expression of the integrin αEβ7, which interacts with MADCAM1, expressed on the endothelium of the intestinal-tissue HEVs, thereby facilitating homing to the mucosal effector compartments. The chemokine CCL25, which is produced by epithelial cells of the small intestine, is also involved in the migration of lymphocytes to the intestinal mucosa, and its receptor, CCR9, is expressed by almost all lymphocytes of the small intestine. The integrin αEβ7 is expressed by IELs and interacts with E-cadherin, which is expressed by the epithelial cells, facilitating tethering of the iels to the intestinal epithelium.
IELs indicates intraepithelial lymphocytes; PPs, Peyer’s patches; MLNs, mesenteric lymph nodes; MADCAM1, mucosal addressin cell-adhesion molecule 1; HEVs, high endothelial vessels; CCL25, cc-chemokine ligand 25.
From Cheroutre H, Madakamutil L. Acquired and natural memory T cells join forces at the mucosal front line. Nat Rev Immunol. Apr 2004;4(4):290-300.3 Adapted by permission from Macmillan Publishers Ltd.
It is believed that after encountering antigen in the inductive compartment, lymphocytes egress into the systemic circulation, where further maturation and differentiation occurs. These differentiated, effector memory (TEM) cells migrate back to the effector sites and await stimulation to discharge effector function.3,10 This is regulated by the coordinated interaction of various cell surface molecules on the T cell with their respective ligands on the surface of vascular endothelial cells in the GI tract. T cells expressing α4β7-integrin11, aEb7-integrin12 and C-C chemokine receptor 9 (CCR9) (Figure 1)13 are perhaps the best characterized in terms of homing potential to the GI tract. The ligand for α4β7-integrin is mucosal vascular–addressin cell-adhesion molecule 1 (MAdCAM-1),11 which is expressed under steady-state conditions by endothelial cells in the GI tract and associated lymphoid tissues.14 Intestinal intraepithelial lymphocytes and a subset of LPLs express aEb7-integrin, the ligand for which is E-cadherin,15 located on intestinal epithelial cells. A subset of circulating α4β7hi T cells selectively express CCR9. The ligand for CCR9, CC-chemokine ligand 25 (CCL-25), is constitutively and selectively expressed by epithelial cells of the small intestine.3,16,17 Expression of GI-specific homing markers is induced following priming of T cells in MLNs and other secondary lymphoid organs draining the GI tract.18,20 Recent data demonstrate that MLN resident dendritic cells are necessary and sufficient in the induction of GI-associated homing receptors.19
| Mechanisms of Intestinal CD4+ T-Cell Depletion | Top of page |
Recent data obtained from the SIV-macaque model provide startling insights into the degree of GI CD4+ T cell infection and depletion as well. The aforementioned studies by Mattapallil32 and Li,33 working with their colleagues, demonstrate rapid infection and destruction of GI memory CD4+ T cells within days of infection with SIV. As mentioned, the former study suggests that, at peak viremia (day 10), as many as 60% of GI memory CD4+ T cells may be infected with SIV32 and that these cells are lost within 14 days of infection. This results in profound immunodeficiency that begins within days of infection, not months to years as was previously thought. The authors put forth the notion that memory CD4+ T cells are killed by direct, virus-mediated destruction rather than by bystander effects or suppression of CD4+ T cell production. Li and coworkers showed, somewhat surprisingly, that GI cells initially infected with SIV have a nonactivated (CD69-CD25-Ki67-) phenotype.33 Peak infection of memory CD4+ T cells within the GI tract corresponded to peak viremia, and depletion of GI CD4+ T cells coincided with a drop in the peripheral viral load. The studies investigators therefore suggested the concept of “substrate depletion” resulting in viral load reduction in the host. In contrast to Mattapallil and coworkers, and as discussed above, the Li study authors propose that CD4+ T cell depletion is secondary to virus-triggered, Fas-Fas ligand mediated apoptosis. Again, it is likely that CD4+ T cell destruction is multifactorial, caused by virus-induced cytolysis, apoptosis, as well as by the host’s own CTL, NK-cell responses.
Picker and coworkers have studied the relationship between mucosal and systemic CD4+ T-cell dynamics and disease progression.40 Rhesus macaques were infected with either CCR5-tropic SIVmac239 or a CXCR4-tropic variant of SIVmac155T3. As expected, those infected with CCR5 tropic virus demonstrated profound depletion of TEM cells associated with initial peak viremia of 107 to 108 RNA copies. Interestingly, this was followed by a prompt proliferative response in the CD4+ T-cell memory population, documented by both Ki67 staining as well as accelerated decay of BrdU labeling. Although macaques infected with CXCR4-tropic virus had markedly high levels of peak viremia and rapid and profound drops in peripheral CD4+ T-cell counts, they were clinically indistinguishable from the remaining macaques. However, a subset of SIVmac239-infected macaques progressed to simian AIDS within 200 days of infection. In these animals, the proliferative burst in the memory CD4+ T-cell population could not be sustained beyond day 42 of infection. It would therefore appear that lack of preservation of mucosal immunity is associated with early disease progression in this model. In addition, it would appear that the ability to maintain the proliferative burst of CD4+ memory T cells, presumably from either the naïve T-cell or central memory population, is a key determinant in SIV pathogenesis.
We have also explored putative virologic and immunologic factors associated with GI CD4+ T cell depletion and conclusively demonstrated, using PCR–based methods, that the HIV-1 viral burden—both proviral DNA and HIV-1 RNA—is consistently greater in the GI CD4+ T-cell population when compared with the peripheral blood CD4+ T cells (Figure 5).41 By combining in situ hybridization and immunohistochemistry, we found both activated and nonactivated cells expressing HIV-1 mRNA at days 18 to 19 postinfection.
Figure 5. HIV-1 Viral Burden is Greater in Gastrointestinal-Derived CD4+ T Cells Compared with Peripheral Blood–Derived CD4+ T Cells in Acute and Early HIV-1 Infection.
GI CD4+ T cells harbor a greater viral burden than PB CD4+ T cells during acute and early HIV-1 infection. PBMC and MMCs from subjects with acute and early HIV-1 infection were sorted via flow cytometry with >99.5% purity. HIV-1 viral DNA and RNA levels were quantified and compared between GI and PB CD4+ T cells.
(A) The log10 HIV-1 viral DNA copy number per 500 CD4+ T cells (shown on the y axis) is compared in 11 study subjects (depicted on the x axis) with red bars representing the PBMC and green bars representing the MMCs.
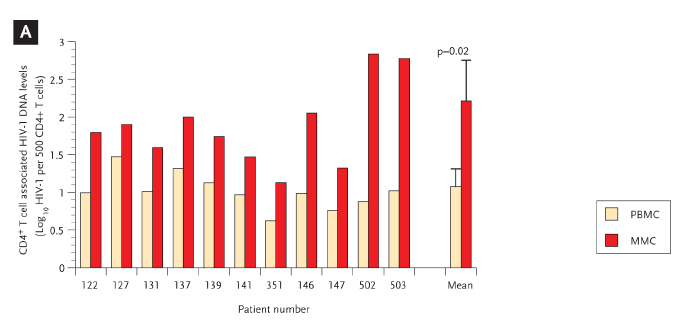
(B) The log10 HIV-1 RNA levels normalized by GAPDH signal (shown on the y axis) are compared in 12 study subjects (represented on the x axis) with red bars depicting the PBMC and green bars depicting the MMCs.
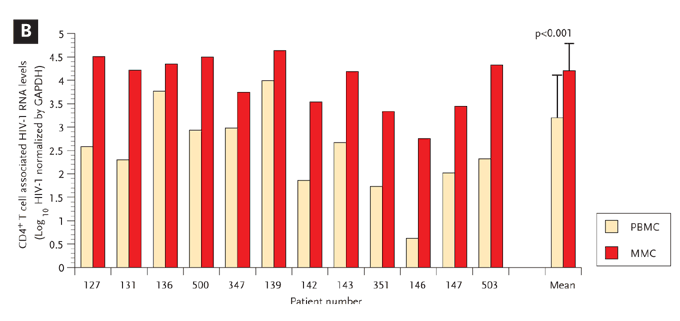
GI indicates gastrointestinal; PB, peripheral blood; PBMC, peripheral blood mononuclear cells; MMC, mucosal mononuclear cells; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
From Mehandru S, Poles MA, Tenner-Racz K, et al. Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J Virol. Jan 2007;81(2):599-612.41 Reprinted with permission from ASM and the author.
Furthermore, high levels of virus in the GI tract were observed to be associated with activation and proliferation of mucosal lymphocytes and a rather dramatic increase in cytotoxic cells, both CD8+ and CD8-. Levels of immune activation, as indicated by CD38 expression on T cells, has long been known to be a major prognostic indicator in patients infected with HIV-1, particularly in the era preceding highly active antiretroviral therapy (HAART).42,43 We have demonstrated that during acute and early infection (AEI), there is a substantial increase in activated memory cells (CD45RO+/CD38+) within the GI tract (Figures 6 and 7). While this, in itself, is not surprising, the observation can have 2 potential consequences.
Figure 6. Significantly Higher Levels of Immune Activation in the Gastrointestinal Tract During Acute and Early HIV-1 Infection Compared with the Levels of Activation in the Peripheral Blood.
(A) Representative flow cytometry plots comparing activated memory cells between an uninfected control (upper panels) and a subject with acute HIV-1 infection (lower panels). PBMCs (left column) and MMCs (right column) were initially identified on the basis of forward and side scatter characteristics. CD3+/CD4+ gated PBMC and MMCs were then analyzed for the expression of CD45RO (x axis) and CD38 (y axis).
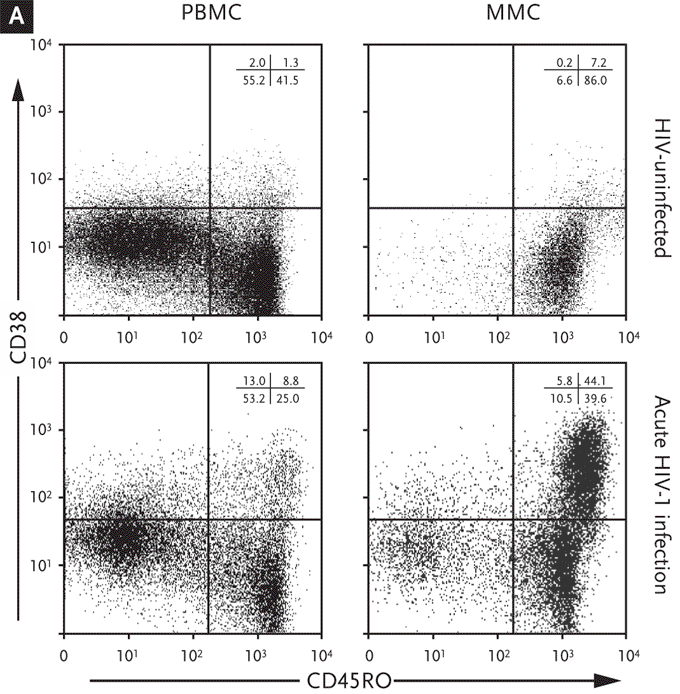
(B) Cumulative data from uninfected controls and subjects with acute and early infection comparing activated memory CD4+ T cells (CD3+/CD4+ gated PBMCs and MMCs coexpressing CD45RO and CD38, depicted on the y axis). This figure shows that, in subjects with acute and early HIV-1 infection, the lymphocytes derived from the GI tract have significantly higher levels of immune activation compared with lymphocytes derived from the peripheral blood.
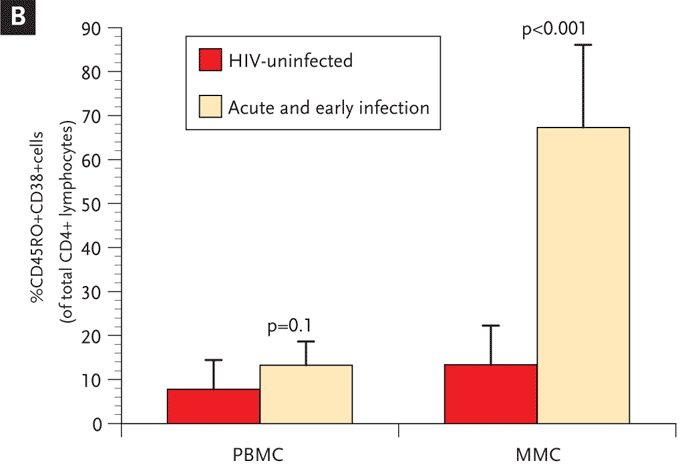
GI indicates gastrointestinal; PBMCs, peripheral blood mononuclear cells; MMCs, mucosal mononuclear cells.
From Mehandru S, Poles MA, Tenner-Racz K, et al. Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J Virol. Jan 2007;81(2):599-612.41 Reprinted with permission from the American Society of Microbiology and the author.
Firstly, most mucosal cells are terminally differentiated effector cells and are much more likely to apoptose when activated than are peripheral blood derived naïve cells. Therefore, given that about two-thirds of mucosal CD4+ T cells express markers of activation (CD38) during acute infection, it is likely that activation-induced cell death plays an important role in GI CD4+ T-cell depletion in this setting. Secondly, activated CD4+ T cells represent the “preferred cellular targets” for HIV-1.44,45 Thus, it would appear that HIV-1 infection generates an expanding population of cellular targets within the GI tract by triggering activation of densely packed mucosal mononuclear cells (Figure 7).
Based on these observations, we have proposed that mucosal CD4+ T-cell depletion during AEI is multifactorial, and is due to (although not limited to) a combination of direct viral infection, activation-induced cell death, and host-derived cytotoxic cellular response. During acute and early infection with HIV-1, GI CD4+ lymphocytes are believed to be preferentially infected and have a greater viral burden compared with PB CD4+ lymphocytes (Figure 7). Although our studies represent snapshots of postpeak viremia events, the fact that up to 102-fold–greater HIV-1 RNA levels were observed in GI CD4+ T cells compared with PB CD4+ T cells is consistent with the concept that the GI tract preferentially supports early events during acute infection. This is likely due to the presence of densely clustered memory CD4+ T cells expressing high levels of CCR5 in the GI tract (Figure 7).26,28
Figure 7. Proposed Model of Immune CD4+ T-cell Depletion in the Gastrointestinal Tract During Acute and Early HIV-1 Infection.
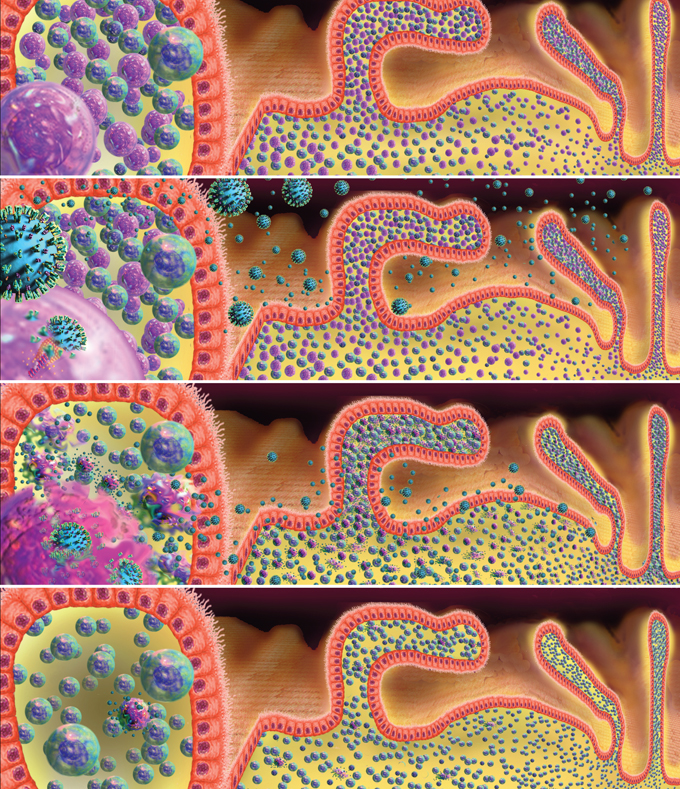
Panel (I) shows a simplified representation of lamina propria lymphocytes separated from the intestinal lumen by a single layer of epithelial cells. CD4+ T cells are shown in pink and CD8+ T cells in blue. We have emphasized dense clustering of lymphocytes in close proximity to the external environment here. Panel (II) shows HIV in the intestinal lumen and lamina propria. On the left side of the panel, a virion fusing with a CD4+ T cell membrane is depicted. In panel (III), lamina propria CD4+ T cells are shown producing virus particles at peak viremia. Panel (IV) shows the sequalae of primary HIV-1 infection with emphasis on the profound loss of intestinal CD4+ T cells.
Images and virus models created by Louis E. Henderson, PhD
| Conclusions | Top of page |
A number of important conclusions emerge from the recent studies examining the GI tract during acute and chronic HIV-1 infection (Table 4):
- First and foremost, it is clear that the GI tract is targeted during all stages of HIV-1, and this is especially so during AEI. We and others have documented that CD4+ T cells are preferentially lost from the GI tract within weeks of HIV and SIV infections. Recent data have demonstrated that GI CD4+T-cell depletion is also observed in natural hosts of SIV—the sooty mangabees and African green monkeys.58,59 Therefore, both pathogenic (HIV-1 infection in man and SIV infection in rhesus macaques) as well as nonpathogenic (SIV infection in sooty mangabees and African green monkeys) lentiviral infections are characterized by depletion of GI CD4+ T cells during acute and early infection. This would suggest that the GI tract is critical in establishment of such infections in a new host. The subsequent role played by the GI tract in disease pathogenesis remains an area of active investigation.
- The mechanisms of GI CD4+ T-cell depletion are probably complex and multifactorial—direct and indirect viral cytopathicity, host-derived cellular immune response, activation-induced cell death and, perhaps, alterations in mucosal homing are all responsible.
- Despite long-term antiretroviral therapy, CD4+T-cell reconstitution remains deficient in the GI tract in spite of the reconstitution seen in the peripheral blood.
- In the short term, patients appear to be asymptomatic. However, vigilance for increased risk of infections, polyps, and malignancies is warranted in the long term, especially as the longevity of HIV-1–infected individuals increases.
- With regard to preventive efforts directed against HIV-1, recent data underscore the need to develop strategies to protect mucosal surfaces from infection. The use of microbicides and CCR5 blockers represents such interventions.
- Finally, and perhaps most importantly, these findings re-emphasize the fact that mucosal immune responses must be targeted in the development of effective HIV-1 vaccines.
| Table 4.Conclusions |
| The GI tract likely plays an important role in the
establishment of lentiviral infections in a new host. The role of the GI tract in disease pathogenesis is unclear and is the subject of active investigation. |
| CD4+ T cells fail to completely reconstitute in the GI tract in a majority of patients despite protracted, apparently suppressive antiretroviral therapy; in contrast, CD4+ T cells reconstitute well in the peripheral blood. |
| Short-term consequences of lack of GI immune reconstitution are not evident, but vigilance is warranted for polyps, cancers, and infections—particularly since the HIV-1 infected population is now surviving over the long term. |
| Protection of mucosal lymphocytes will likely be critical in HIV-1 vaccine and microbicide development efforts. |
| GI indicates gastrointestinal |
| Grant Support | Top of page |
This work was supported in part by the following grants: The Acute HIV Infection and Early Disease Research Program (AIEDRP) (AI-41534) (PI: M. Markowitz) and the American Foundation for AIDS Research (AMFAR) (106717-40-RGRL) with support from Concerned Parents for AIDS (CPFA) (PI: M. Markowitz), the General Clinical Research Center grant from the National Center for Research Resources at the National Institutes of Health (M01-RR00102), and the German Ministry of Education and Research (BMBF) Contract KompNet 01KI0211 (Paul Racz and Klara Tenner-Racz).
| Acknowledgements | Top of page |
We would like to thank the patients for their participation. We acknowledge the nursing staff at Rockefeller University and Bellevue Hospitals for their clinical assistance. Further, we would like to thank Petra Meyer, Birgit Raschdorff and Gudrun Großschupff for their technical assistance with immunohistochemistry.
| References | Top of page |