
There is still considerable controversy over appropriate clinical management strategies for patients diagnosed with HIV during acute or early stage infection. However, it is widely accepted that enrolling longitudinal cohorts to study such patients is vital. Long-term outcome studies of persons enrolled early in the course of infection continue to yield important data regarding transmission dynamics, disease progression, efficacy of treatment, and incidence and consequence of superinfection. Not only are these studies helping to determine how best to clinically manage patients diagnosed early in the course of HIV disease, they are contributing significantly to the research exploring virologic, immunologic, and treatment questions in patients with chronic infection.
The Options Project, developed and conducted by the University of California, San Francisco (UCSF) AIDS Program at San Francisco General Hospital, is dedicated to the study of acute and early HIV infection. Dr. Frederick Hecht, a co-director of the program, and his colleagues continue to make headway in exploring various pathogenesis- and treatment-related issues affecting newly infected patients. In October, Dr. Hecht returned to PRN to provide members with an update on key data generated by the project, along with the recent findings from Acute Infection and Early Disease Research Program (AIEDRP), a national, multisite network consisting of research centers actively recruiting patients in the early stages of HIV infection. Here we present the highlights of his comprehensive talk.
| I. Treatment and Treatment Interruptions in Early HIV Infection | Top of page |
The interest in studies of structured treatment interruptions (STIs)-as a component of antiretroviral therapy started during acute or early HIV infection-dates back to a preliminary evaluation conducted by Dr. Eric Rosenberg and his colleagues at Massachusetts General Hospital (Rosenberg, 2000). This small study followed eight patients who began therapy during acute HIV infection and agreed to initiate STI after successful viral suppression, with the plan to restart therapy if viral load exceeded 5,000 copies/mL for three consecutive weeks or exceeded 50,000 copies/mL at any given time. Despite rebound in viremia, all eight patients were able to achieve at least a transient steady-state off therapy with viral loads below 5,000 copies/mL. At the time the report was prepared for publication in Nature, five out of eight subjects remained off therapy with viral loads less than 500 copies/mL after a median of six-and-a-half months. Dr. Rosenberg's group observed increased virus-specific CTLs and maintained HIV-specific CD4+ cells in all eight patients.
More recently, the Massachusetts General group reported extended follow-up data regarding six of the original subjects, along with eight additional patients who underwent a similar STI process (Kaufman, 2004). All 14 patients were HIV-antibody negative or in early seroconversion and had high HIV-RNA levels prior to beginning treatment (all patients received either a nelfinavir [Viracept]- or indinavir [Crixivan]-based regimen). Prior to initiating the first of up to four STIs, patients maintained HIV-RNA levels below 50 copies/mL for at least eight months while on treatment. And much like the eight-patient pilot study, patients were required to restart treatment if their viral load exceeded 5,000 copies/mL for three consecutive weeks or if they viral load exceeded 50,000 copies/mL at any one time.
The 14 patients in this STI study were followed for a median of 5.3 years. Eleven of the 14 (79%) patients were able to achieve viral loads of less than 5,000 copies/mL for at least 90 days following one, two, or three interruptions of treatment. However, a gradual increase in viremia and decline in CD4+ cell counts was observed in most individuals. By an intent-to-treat analysis, eight (57%), six (43%), and three (21%) of the 14 patients achieved a maximal period of control of 180, 360, and 720 days respectively, despite augmentation of HIV-specific CD4+ and CD8+ cell responses (see Figure 1). The magnitude of HIV-specific cellular immune responses before treatment interruption did not predict the duration of virologic control. The study authors also commented that the small sample size, as well as the lack of concurrent untreated controls, does not allow for the assessment of possible clinical benefit using this approach.
Figure 1. Mass General Cohort: Follow-Up of 14 Patients
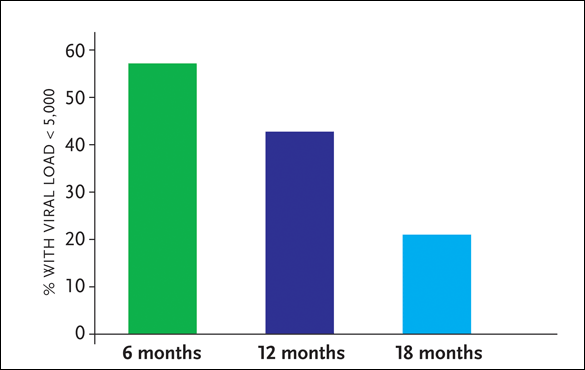
Source:Kaufman, 2004
“While these data suggest that STIs after treatment of primary HIV infection do not yield the level of dramatic control of viral replication suggested by the initial report, they raise some important questions,” Dr. Hecht said. “First, why isn’t viral control maintained? Second, is there a lower, but less dramatic difference in viral load set point? Figuring out what’s going on, even on a cellular level, could be valuable.”
| III. HIV Superinfection | Top of page |
HIV superinfection—sequential infection with variants of HIV—has long been a source of debate and concern among clinicians and public health officials. The topic is of particular interest to researchers involving in the study of primary HIV infection, given that the five case reports published to date have involving individuals who were recently infected or intermittently treated with antiretroviral therapy (Ramos, 2002; Jost, 2002; Altfeld, 2002; Koelsch, 2003; Smith 2004; Brenner, 2004). Studies of chronically infected patients have not shown evidence of superinfection (Gonzales, 2003; Grant, 2004; Tsui, 2004).
“The fact that we’ve only seen case reports involving newly infected patients suggests that superinfection can occur before development of protective immunity,” Dr. Hecht commented. “Understanding superinfection may be a key to understanding protective immunity and vaccine development. And it most definitely raises patient counseling issues.”
At the 12th CROI in Boston, Dr. Hecht’s group—which included Dr. Robert Grant and his colleagues with the UCSF Laboratory of Clinical Virology—presented data on the frequency of sequentially expressed dual HIV infections (SEDIs) in the Options Project Cohort (Grant, 2005).
Dr. Grant’s lab evaluated samples collected from all patients enrolled in the cohort who had been followed for at least 48 weeks and had remained off treatment. “We first looked at the pol gene—using the sequencing performed when looking for drug resistance—and compared their baseline specimen with their most recent available follow-up specimen,” Dr. Hecht explained. “We conducted phylogenetic analyses to see if the subsequent viruses matched up with the viruses they first had. When it looked like we had a possible superinfection case, we did a lot of retesting with specimens throughout the entire series of time points to see if we could support this finding and to look for exactly when superinfection occurred.” Finally, to evaluate population dynamics, Dr. Hecht’s group used heteroduplex assays to determine if the second virus was evident in the baseline specimens (this method has been validated at 1.5% to 3.0% sensitivity for minor variants). “We can get down to pretty low quantities using these methods to look for evidence of the second virus at early time points.”
A total of 104 recently infected individuals were analyzed at two or more time points, representing 195 person-years of observation. All viral sequences were subtype B. Highly divergent viral sequences appeared in a total of four cases over time, representing an incidence density of 2.1/100 person years. “This incidence is close to what you might expect in a reasonably high-risk cohort in San Francisco of HIV-negative individuals becoming infected with HIV,” Dr. Hecht said. The heteroduplex assays confirmed the lack of second virus at baseline, indicating that these were sequential infections.
One patient discussed by Dr. Hecht—provided with the pseudonym “Hector”—saw a highly divergent virus appear 16 to 44 weeks after infection (see Figure 2). “Looking at his HIV-RNA levels, he appeared to be doing very well,” Dr. Hecht pointed out. “Around week 24, his viral load leveled off at approximately 100 copies/mL. Then, around week 40, there was a sharp increase in his viral load, spiking at around 10,000 copies/mL. It was here that we noted that he had a different virus than the one he started off with. His CD4+ cell count, which was initially doing well, started to decline rapidly. In fact, he had to go on treatment within another few months of follow-up.”
Figure 2. "Hector"
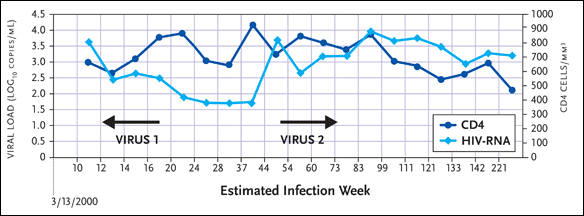
Source:Schweighardt, 2005
Another interesting case is “Cody” (see Figure 3). A sharp increase in his viral load occurred between weeks 43 and 53, before his second virus was detected. In this patient, multiple-drug-resistant virus was replaced by wild-type virus, which was associated with more rapid disease progression. His CD4+ count, reaching a high of approximately 900 cells/mm3 at week 35, fell to a low of approximately 300 cells/mm3 at week 79.
Figure 3. "Cody": Replacement of MDR HIV with Wild-Type HIV
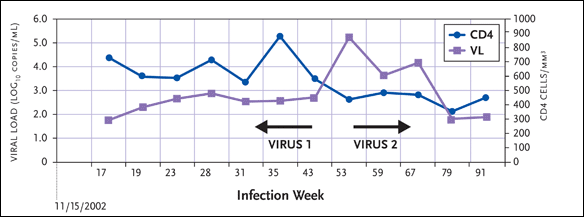
Source:Schweighardt, 2005
With “Jonah,” the second virus appeared concomitantly with a diagnosis of gonorrhea, at week 168 (see Figure 4). “His viral load spiked at 10,000 copies/mL at the time of the gonorrhea diagnosis and his CD4+ cell count began a rapid decline after this time point.”
Figure 4. "Jonah": Virus 2 Appears with Diagnosis of Gonorrhea
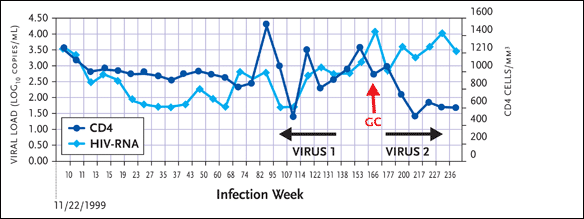
Source:Schweighardt, 2005
“Leon’s” first virus was detected through to week 29. His second virus was first detected at week 39. Around the time his second virus was detected, his CD4+ count was approximately 700 cells/mm3. Around week 48, his CD4+ count plummeted to 350 cells/mm3. After a transient increase, his CD4+ count fell below 100 cells/mm3 around week 100. “With this patient, the second virus appeared before rapid progression to AIDS.”
“All four patients saw increases in their viral loads with their second viruses,” Dr. Hecht said. “The average increase was 1.4 log10 copies/mL, compared to an average decrease of 0.24 log10 copies/mL in our untreated cohort participants without superinfection.”
In summarizing these data, Dr. Hecht remarked that superinfection appears to be relatively common in early HIV and may be associated with increased viral load and more rapid disease progression. “What we’re interested in finding out about now is the closing of the superinfection window. Why do people stop becoming superinfected, at least from what we think we’re seeing, after the first few years of infection? This question remains unanswered.”
| References | Top of page |