
Patients who have developed resistance to the 3 currently available classes of HIV medications present a particular treatment challenge. Developments in an entirely new class of HIV antiretroviral therapy, inhibitors of the viral integrase protein, were the focus of discussions at a recent PRN meeting.
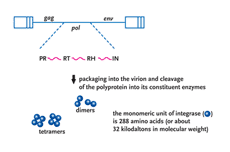
HIV-1 integrase is a viral protein that has several roles (Figure 1). After the completion of reverse transcription of the viral RNA genome into DNA, it appears in part, to be responsible for ferrying the viral DNA into the nucleus of the cell, a position where it has the ability to closely associate with one of the chromosomes of the host cell. Subsequently, integrase catalyzes the insertion, or integration, of viral DNA into the host cellular DNA, a step of the viral lifecycle required for reprogramming the cell to produce more copies of itself (Goff, 1992; Kulkosky, 1994; Lewinski, 2005). Integration is, in effect, the most intimate association any virus can make with its host the irreversible chemical fusion of its DNA with that of the cell. Chromosomal DNA then serves as a sanctuary for the expression of viral information as well as the perpetuation of the retroviral genome. Once integrated into the host chromosome, retroviral DNA is a permanent fixture and upon cell division, is passively transmitted as an embedded passenger within the DNA of all daughter cells. Thus, the infected cell is a predisposed source of virus production not only for the lifetime of that cell but for all subsequent generational derivatives (reviewed in Coffin, 1997).
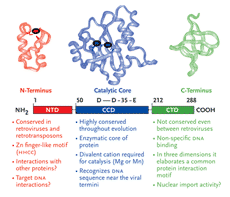
HIV-1 integrase is a member of a protein superfamily known as the polynucleotidyl transferases (Mizuuchi,1997) (also see sidebar on page 10). It has an ancient and highly conserved catalytic domain, described not only in human retroviruses but also found in the retroviral-like elements of the fruit fly, Drosophila and yeast (Flavell, 1997), and even exists in certain primitive transposons found in bacteria (Hallet and Sherratt, 1997; Polard, 1995; Rice,1996). Perhaps paradoxically, the formation of the human immune system also requires a similar functional domain as a part of the RAG recombinase system (Jones, 2004; Schatz, 2004). The RAG enzymes are responsible for the maturation of B and T cells, which in turn, are critical to fight against invading microorganisms, like HIV-1.
In the context of the HIV lifecycle, the integration event itself divides the viral life cycle into 2 distinct phases (preintegration and postintegration)
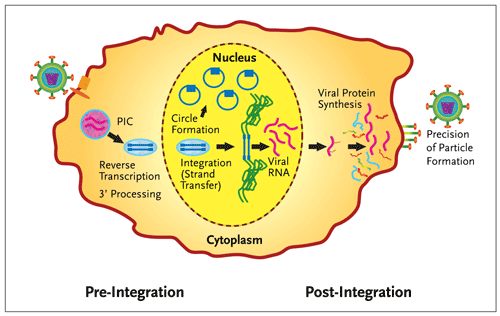
Figure 1.Integrase Divides the Retroviral Lifecycle.
Integration of HIV-1 DNA (blue) into the host chromosome (green) divides the retroviral life cycle. In addition to the catalytic functions required for integration (3 processing and strand transfer), integrase is also involved in other essential functions (reverse transcription, the circularization of the viral DNA into extra-chromosomal forms and the precision of virion particle formation). PIC is the preintegration complex, an operationally defined association of viral and host proteins that coordinate reverse transcription and nuclear import of the viral genome. In this figure, viral entry is on the left and egress of newly formed particles on the right.
However, there are other steps either preceding or immediately after the step of chemical bonding between the viral and host DNAs that are equally essential for the completion of this complicated set of enzymatic reactions. First, while still in the cytoplasm, integrase cleaves 2 nucleotides from the ends of newly reverse transcribed and linear viral DNA. This reaction (the 3' processing reaction) generates a reactive chemical group (3' hydroxyl) at each terminus of the viral DNA (Figure 2).
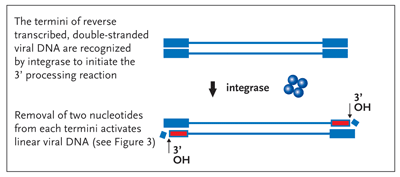
Figure 2. The 3Processing Reaction Activation of the Viral DNA Termini
Following the completion of reverse transcription of the viral RNA into its DNA copy, integrase (here shown as blue tetramer) removes two nucleotides from the 3 end of each strand of the viral DNA generating a chemically reactive hydroxyl group (3OH, red). This reaction serves to activate the termini of the linear dna molecule for a subsequent strand-transfer step outlined in Figure 3, the target of all integrase inhibitors currently in the clinic.
Integrase is then imported into the cell nucleus by an unknown mechanism within the context of a preintegration complex, or PIC. The PIC is composed of viral and host proteins in conjunction with the viral DNA. The nuclear (viral) DNA then undergoes one of two fates. In the strand-transfer reaction (Figure 3), the activated 3' hydroxyl groups left after the 3' processing reaction attack the host chromosome on opposite sides of the DNA helix, producing a 5-base-pair staggered cut, simultaneously forming stable chemical bonds between the inserted viral DNA and that of host cell DNA. Finally, the ragged ends produced on opposing sides of the sealed recombinant joint are repaired by as yet unknown host factors and the integrity of the chromosomal DNA restored.

Figure 3. The Strand Transfer Reaction Intimate and Irreversible Bonding to Host Cell DNA
Integrase utilizes the reactive chemical groups produced in Figure 2 (red, 3 OH) to simultaneously make a 5-base-pair staggered cut in the host chromosomal DNA (green) and then to stably introduce the viral DNA at the cleaved sites. This occurs as a concerted reaction and is reversible in vitro. However, once the integrity of the chromosomal DNA is restored by the repair of the recombinant joint by the action of undefined host factors (man-at-work sign), the viral DNA insertion becomes indistinguishable from the rest of the cellular dna and cannot be removed.
It is amazing that the integrated viral DNA copy, comprising only about 0.0003% of the entire genetic capacity of the cell, can now commandeer the cellular environment to its own advantage. The other fate of nuclear viral DNA is the circularization of these linear molecules into extrachromosomal forms (Figure 4). They are the products of the activities of cellular enzymes in the nucleus. Although the function of the circles with regard to their contribution to the viral lifecycle is not yet understood, it has been known for some time that these molecules can act as templates for limited expression of the viral genome (although clearly not enough to produce all of the viral proteins necessary to assemble the mature virus) (Wiskerchen, 1995; Wu, 2001; Wu, 2003). Whether or not the low level of viral gene expression originating from the circles contributes to the pathogenesis of the virus remains to be determined.
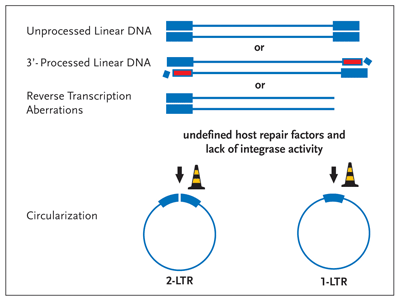
Figure 4. Circularization— An Alternative Fate of Linear Viral DNA
Linear viral DNA is circularized by host nuclear proteins in the absence of integrase enzymatic activity. If integrase fails to process the 3 termini (Figure 2) or if the enzyme fails to perform the strand transfer reaction (Figure 3), the viral DNA is circularized by the activity of still ill-defined host proteins (road work cones). Although these circular DNA molecules can act to produce a subset of viral proteins, they can't provide for the synthesis of the viral structural gene products, key to virion formation and infectivity
| Therapeutic Discoveries | Top of page |
In light of the inability of HIV to propagate itself without integration of its genome into chromosomal DNA, the viral integrase protein has become an important potential therapeutic target. It is distinct mechanistically, independent of the catalytic activities of the viral protease, reverse transcriptase, or of virus cell fusion. It therefore has the potential for synergistic activity in combination with available protease and reverse transcriptase inhibitors, and with entry inhibitors now in development. In addition, because of their novel molecular structure, the cost of production of the new families of integrase inhibitors may be less than that of some of the current antiretrovirals which rely on protein-like compounds or nucleoside derivatives for their activity. Although it has been more than a decade since integrase was first described, it has been very difficult to find therapeutic inhibitors. For instance, the integrase protein is insoluble in its native form and it has been relatively difficult to purify and study its biochemical and physical properties. It is anticipated that, given the properties of the enzyme, it may well be that several forms of the protein may exist, perhaps with each structural configuration relevant to a specific activity of the protein. The search for new inhibitors has also been confounded by the fact that many of the multiple compounds that have been described as inhibiting the activities of integrase in vitro fail to exhibit specific activity against integration during viral infection in vivo. Indeed, very few of the lead compounds found in vitro can satisfy the 4 critical qualifications that were recently outlined (Pommier, 2005). These criteria are: 1) time of drug addition experiments that must show that the drug is active after the completion of reverse transcription (about 4-6 hours postinfection [hpi]) but not after 10 to 12 hpi, the approximate timing of HIV-1 integration; 2) there must be a concomitant rise in the accumulation of circular species in treated cells indicating that the drug specifically inhibits HIV integration allowing for the alternative fate of viral DNA (circularization) to predominate; 3) the location of mutations associated with drug resistance must map to integrase; and 4) purified mutant integrase protein must exhibit associated drug resistance in standard biochemical assays for the strand transfer reaction. Of great assistance to the development of potential compounds was the assay system to detect the specific inhibition of the integrase-catalyzed strand transfer reaction (Hazuda, 2000). Among the inhibitors screened for activity, a set was found that led to the specific inhibition of HIV integration (the strand transfer reaction) and interrupted replication in tissue culture cells.
Subsequently, Dr. Hazuda and colleagues published the result of a proof-of-principle trial that demonstrated that compound L-870,810, a naphthyridine carboxamide could inhibit retroviral replication in rhesus macaques (Hazuda, 2004). Other groups have also developed potential drugs, most of which are thought to act by inhibiting strand transfer. No compounds that inhibit the 3' hydroxyl processing reaction are in human clinical trials. Merck-0518 and Gilead-9137 are currently being evaluated in the clinic. They are active in the nanomolar range, their activity generated, in part, from bond formation between metal ions contained at the active site of the enzyme and the compound (Grobler, 2002). Mutations originating from drug resistance studies of similar integrase inhibitors cluster near a region in the 3-dimensional structure near the active site and overlapping a region of residues that appear to bind DNA. Interestingly, there may be a requirement for drug binding in the context of a ternary complex, which means that the drug might interface not only with integrase, but also with the cations and nucleic acids that are present at the active site.
| References | Top of page |

{kind=link}
{kind=link}