
| Introduction | Top of page |
Constructing effective salvage therapy regimens for HIV-infected patients with highly resistant virus poses a daunting challenge to the practicing clinician. The recent approval of new protease inhibitors (PIs)—tipranavir and darunavir—and the continuing development of new drug classes provide hope that patients can now be salvaged with at least a few active new drugs. This paper reviews the available clinical outcomes data for tipranavir and darunavir as well as the investigational CCR5 antagonists, 2nd-generation nonnucleoside reverse transcriptase inhibitors (NNRTIs), and integrase inhibitors. Also discussed are salvage strategies in treatment-experienced patients with the goal of maximizing the chances of a patient receiving at least 2 active agents in combination. When this is not possible, the continued use of nonsuppressive therapy through de-escalation to a simplified “holding” regimen may be utilized to maintain less fit hiv mutants until more suppressive treatment options become available.
| Recently Approved Protease Inhibitors | Top of page |
Tipranavir
Approved in 2005, tipranavir (TPV; Aptivius®) is a novel nonpeptidic PI with demonstrated activity against PI-resistant HIV. It is supplied as 250 mg soft gel capsules and is dosed at 500 mg twice daily and coadministered with 200 mg of ritonavir twice daily. This increased dose of ritonavir has led to concerns of increased adverse effects and toxicity. In vitro data suggest that tipranavir lowers ritonavir drug levels and could ameliorate the effects of increased ritonavir dosing (Sabo, 2006). However, gastrointestinal system disorders were more frequently seen in subjects receiving tipranavir-ritonavir than a comparator PI (CPI)- ritonavir in a combined analysis of 2 phase 3 trials that formed the basis for tipranavir’s approval (Hicks, 2006). These 2 trials, known as the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies, compared the safety and efficacy of tipranavir-ritonavir plus an optimized background regimen (OBR) with a ritonavir-boosted CPI plus OBR. Roughly 50% of subjects randomized to a comparator PI received lopinavir/ritonavir. Enrolled subjects had a baseline viral load of 4.73 log10 copies per mL and a cd4 count of 196. Approximately 20% of subjects in each arm entered the studies with a cd4 count below 50. The study populations were heavily treatment experienced with median prior treatment use of 6 nucleoside reverse transcriptase inhibitors (NRTIs), 1 NNRTI and 4 PIs. Subjects were excluded if they had 2 or more PI mutations at positions 33, 83, 84 and 90. Ten percent of subjects had previously used enfuvirtide. At 48 weeks, subjects receiving boosted tipranavir demonstrated greater mean viral load reductions (−1.14 log10 copies/mL v −0.54 log10 copies/mL) and were more likely to maintain a greater than 1 log10 viral load reduction from baseline (33.6% vs 15.3%) compared with subjects receiving cpiritonavir. At 96 weeks, a greater proportion of subjects in the boosted tipranavir arm achieved a viral load below 50 copies than did those in the comparator PI arm (20.4% vs 9.1%) (Farthing, 2006). Figure 1 illustrates that the likelihood of achieving a treatment response or a viral load below 50 copies at 48 weeks was significantly influenced by the use of enfuvirtide (P<.0001 for both) (Cahn, 2005). These data underscore the important point that salvage therapy is best undertaken when at least 2 active drugs are available.
Figure 1. The influence of enfuvirtide (ENF) use on virologic outcomes in subjects enrolled in RESIST 1 and 2. (A) Treatment response as a function of tipranavir-ritonavir/enfuvirtide co-administration. Treatment response is defined as ≥ log10 viral load reduction without treatment change at 48 weeks. (B) Percentages of subjects with an undetectable viral load (<50 copies/mL) at 96 weeks subdivided by first-time enfuvirtide use.
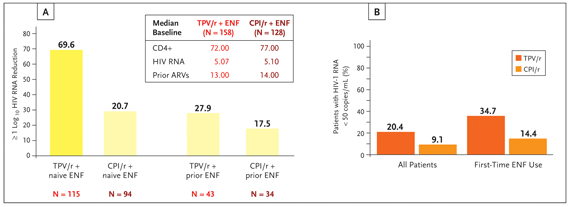
Source: Reprinted with permission from Cahn P, Hicks C, for the resist study teams. Paper presented at: X European aids Conference; November 17-20, 2005; Dublin, Ireland. Abstract PS3/8.
Figure 2. Virologic response in darunavir/ritonavir 600/100 mg bid patients with (A) ≥1.0 log10 viral load reduction, and (B) plasma viral load <50 copies/mL from baseline at week 24 [intent to treat (ITT) time to loss of virologic response (TLOVR)].
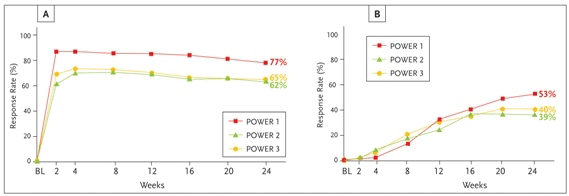
Source: Reprinted with permission from Molina JM, Cohen C, Katlama C, et al. 16th International aids Conference; Toronto, Canada; August 13–18, 2006. Abstract TUPE0060.
Notable grade 3 to 4 laboratory abnormalities, including elevated transaminases and hypertriglyceridemia, occurred more frequently in subjects receiving boosted tipranavir than comparator pi. A warning was issued in June of 2006 alerting clinicians to the increased risk of intracranial hemorrhage in patients taking tipranavir. In most cases, these patients either had comorbid medical conditions or were receiving other medications that may have contributed to the increased risk of bleeding. Tipranavir has important drug interactions with other anti-retroviral agents. It reduces the area under the curve (AUC) for lopinavir, saquinavir and amprenavir meaning that tipranavir should not be used as part of a double-boosted pi regimen. Levels of the investigational 2nd generation nnrti etravirine (formerly TMC-125) are decreased by tipranavir; they should not be coadministered.
Figure 3. Percentage of patients enrolled in the power and resist studies achieving undetectable viral loads (HIV RNA <50 copies/mL) comparing all patients to those also using enf for the first time.
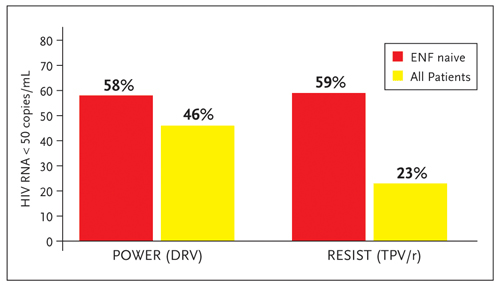
Source: Adapted from Hill A, Moyle G. Paper presented at: 46th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), September 27-30, 2006, San Francisco, California. Abstract H-1386.
Darunavir
Darunavir (DRV, formerly TMC 114; Prezista™) received fda approval in June of 2006 and is indicated for use in treatment-experienced patients. It is supplied as a 300-mg tablet and requires boosting with ritonavir. The recommended tipranavir dose is 600 mg with 100 mg of ritonavir twice daily. Studies exploring a once-daily dosing regimen with darunavir 800 mg and ritonavir 100 mg in ARV-naive patients are ongoing. Darunavir has fewer drug interactions than tipranavir and can be used in conjunction with atazanavir, efavirenz and etravirine. It is not recommended for use, however, with Kaletra®, saquinavir or pravastatin. Approval by the FDA was based on results from the Performance Of TMC114/r When Evaluated in triple-class-experienced patients with PI Resistance (POWER) studies. The POWER studies included triple class resistant subjects failing a PI-based regimen with viral loads above 1000 and with at least 1 major pi mutation. Subjects in power 1 and 2 were then randomized to an OBR plus either a CPI or 1 of 3 dosing regimens of boosted darunavir. POWER 3 contained only 1 darunavir dosing arm at the current FDA-approved twice daily dose. Enrolled subjects had, on average, 3 primary PI resistance mutations and an at least 80-fold baseline change in lopinavir susceptibility. At 24 weeks, subjects receiving twice-daily darunavir 600 mg plus ritonavir 100 mg were more likely to achieve a viral load below 400 copies per mL than subjects receiving comparator PI (63% vs 19%). Virologic response in all 3 POWER studies was similar at the FDA-approved dose (Figure 2). Darunavir was well tolerated with hepatotoxicity and lipid abnormalities equal to or less than currently approved PIs.
Although it is difficult to compare results across studies, Figure 3 summarizes the percentage of subjects achieving hiv rna levels below 50 copies in both the POWER and resist studies at 48 weeks. The 24 week data suggested a greater treatment benefit in enfuvirtide-naive patients treated with darunavir than tipranavir for the endpoints of ≥1 log10 reduction in HIV RNA and HIV RNA <50 copies/mL (Hill, 2006a). By 48 weeks, these differences in enfuvirtide-naive patients were less evident. For all enrolled patients at 48 weeks, the benefit of boosted darunavir over comparator PI was greater than the benefit of boosted tipranavir for the endpoints of ≥1 log10 reduction in HIV RNA and cd4 cell count increase (Hill, 2006b). However, the statistical significance of these reported differences remains to be determined.
Figure 4. Tipranavir- and Darunavir-associated mutations. Mutations conferring resistance to both drugs are shaded in gold.
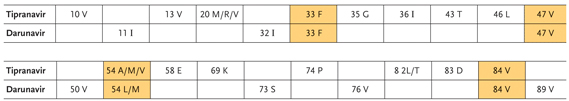
| Conclusion | Top of page |
The availability of new PIs, NNRTIs, and investigational agents from newer classes increases the likelihood that complete virologic suppression can be achieved in an increasing proportion of triple class-resistant patients. Three drugs from previous classes, tipranavir, darunavir and etravirine, and 3 drugs from novel classes, enfuvirtide, maraviroc, and Merck-0518 should all be available over the next year for use in salvage regimens. Sequential monotherapy should be avoided. If possible, clinicians would be wise to wait until there are enough agents available to combine for a given patient and produce a high probability of achieving both a durable response and a viral load below 50 copies per mL.
| References | Top of page |