
There are 20 unique medications approved for the treatment of HIV infection. Despite this impressive number, there is an indisputable need for new anti-HIV compounds that have potent and durable efficacy profiles, unique resistance patterns, patient-friendly dosing schedules, and minimal toxicities. What follows is an overview of some the newest antiretroviral contenders, discussed by Dr. Roy M. Gulick during a recent meeting of the Physicians’ Research Network.
| I. Nucleoside/Nucleotide Analogues | Top of page |
D-D4FC (Reverset)
Now in phase II clinical trials is D-D4FC (Reverset), a cytidine analogue being developed by Pharmasset Pharmaceuticals and Incyte Corporation. Reverset’s chemical name is D-D4FC and was formerly known as DPC-817 when it was being developed by DuPont Pharmaceuticals and Bristol-Myers Squibb. It has an intracellular triphosphate half-life of 17 hours, rending it suitable for once-daily dosing.
According to encouraging in vitro data, D-D4FC is highly effective in inhibiting subsets of lamivudine-, zidovudine-, and tenofovir-resistant HIV variants but is less potent against variants harboring multiple-NRTI-resistant mutations. D-D4FC selects, in vitro, for the K65R or K70R mutations, which decreases susceptibility to the drug.
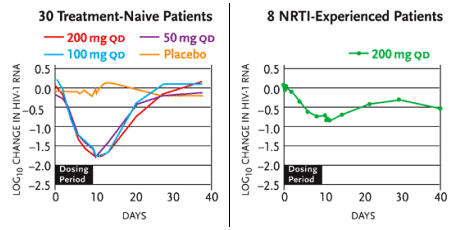
Results of a Phase Ib/IIa study presented by Dr. Robert Murphy of Northwestern University and his colleagues were presented at the XV International AIDS Conference in Bangkok (Murphy, 2004). The placebo-controlled study (Study 202) involved 30 treatment-naïve and 10 treatment-experienced patients. The treatment-naïve patients received 50 mg, 100 mg, or 200 mg D-D4FC monotherapy—or matching placebo—for ten days. Eight treatment-experienced patients, none of whom were maintaining undetectable HIV-RNA levels while taking standard regimens, added 200 mg D-D4FC to their therapies for ten days (and two other others added D-D4FC placebos).
The mean reduction in viral load among the treatment-naïve patients in the three treatment groups ranged from 1.67 log10 copies/mL to 1.77 log10 copies/mL (see Figure 1). Among the treatment-experienced patients, the mean reduction in viral load was 0.8 log10 copies/mL. Seven of eight treatment-naïve patients (in the 200 mg D-D4FC group) and four of eight treatment-experienced patients achieved undetectable viral loads (<400 copies/mL) after ten days of therapy. The drug was well tolerated. No new resistance mutations developed during the ten days of therapy.
Results from a Phase IIb clinical trial (Study 203), involving 180 treatment-experienced patients, are expected soon. Depending on the results of this study, a Phase III clinical trial is planned to begin before the end of 2005.
| IV. Entry and Fusion Inhibitors | Top of page |
Maraviroc
UK-427,857, which now carries the generic name maraviroc, is a chemokine receptor antagonist designed to block the CCR5 receptor. The drug is being developed by Pfizer Pharmaceuticals.
Some preliminary pharmacokinetics data are available (Muirhead, 2005). Maraviroc is a CYP 3A4 substrate, not an inhibitor. In single dose studies in HIV-infected individuals, efavirenz decreased maraviroc by 40% to 50%, whereas nevirapine increased the maraviroc Cmax 1.5 times, with little effect on the maraviroc AUC. Lopinavir/r also increased the maraviroc Cmax approximately 1.8 times.
Preliminary results of a phase I study were presented at the 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy in 2003 (Pozniak, 2003). In this study, 24 HIV-positive individuals with CCR5-tropic virus were randomized to receive once-daily oral doses maraviroc (25 mg QD or 100 mg BID) or placebo monotherapy for 14 days. Steady-state drug levels were achieved after seven days, with the best plasma concentrations achieved when taken in the fasted state. After 14 days, an average 1.4 log10 copies/mL reduction in HIV-RNA was seen in patients receiving maraviroc 100 mg BID, compared to a 0.4 log10 copies/mL reduction among patients receiving the 25 mg QD dose. The drug was well tolerated; no serious adverse events were reported.
The preliminary results of a dose-ranging study were reported at the XV International AIDS Conference in Bangkok (Fätkenheuer, 2004). In this study, 80 patients infected with CCR5-tropic HIV received one of several doses of maraviroc (25, 100, or 300 mg QD; or 50, 100, 150, or 300 mg BID) or placebo for a 10-day period. The agent was well tolerated and caused dose-related reductions in viral load. Using the 100 and 300 mg BID dosing, viral load was reduced by 1.4 to 1.6 log10 copies/mL. Food intake did not affect drug levels or antiviral activity.
Also of interest, the study noted that the CCR5 receptor was saturated by at least 85% with all but the lowest dose studied. With respect to tropism, 61/63 evaluable patients remained CCR5 tropic throughout the 10-day treatment course and throughout the additional 30-day follow-up period. Two patients became dual tropic. One reverted back to CCR5 at day 40, whereas the other patient continued to have a dual-tropic HIV genotype without obvious clinical progression after six months.
Vicriviroc
Vicriviroc is the tentative generic name for Schering-Plough’s CCR5 receptor antagonist, also known as SCH 417690 and formerly known as SCH-D. Like maraviroc, the drug is orally bioavailable. It has a long half-life allowing for once-daily dosing. Concentrations of vicriviroc are enhanced with ritonavir, whereas concentrations are decreased with efavirenz or nevirapine.
Phase I clinical trial results, involving 48 HIV-infected patients with CCR5-tropic virus receiving various doses of the drug, were reported at the 11th CROI (Schurmann, 2004). Sixteen patients were randomized into each once-daily dosing group (10 mg, 25 mg, 50 mg, or placebo) evaluating oral SCH 417690 monotherapy for 14 days (four patients in each dosing group received placebo). All patients were either naïve to antiretroviral therapy or had been off treatment for at least eight weeks.
The drug was well tolerated and no adverse events were documented. After 14 days, HIV-RNA levels were 1.08 log10 copies/mL below baseline in the 10 mg group, 1.56 log10 copies/mL below baseline in the 25 mg group, and 1.62 log10 copies/mL below baseline in the 50 mg group. Approximately 45% of patients in both the 25 mg and 50 mg groups saw their viral loads drop by at least 1.5 log10 copies/mL.
ACTG 5211, a phase II study of vicriviroc with ritonavir and optimized background antiretrovirals in treatment-experienced patients, is actively enrolling patients.
GW 873140
Yet another CCR5 receptor antagonist in development is GlaxoSmithKline’s GW 873140. The drug has a long half-life and 50% occupancy of the CCR5 receptor persists five days after the last dose—with concurrent undetectable plasma drug levels—indicating that the drug likely has a lingering (“post-antiviral”) effect after it is stopped (Sparks, 2005). And like the other CCR5 antagonists, GW 873140 is a CYP 3A4 substrate. Coadministration with lopinavir/r results in a sixfold to eightfold increase in GW 873140 levels and a 30% increase in ritonavir levels (Adkison, 2005).
The results of a ten-day GW 874140 monotherapy study have been reported (Lalezari, 2004). The study enrolled 40 treatment-naïve and treatment-experienced HIV-infected subjects with R5-tropic virus, a CD4+ count above 200 cells/mm3, a viral load above 5,000 copies/mL, and had been off antiretroviral therapy for at least 12 weeks. The patients were randomized to one of four cohorts: 200 mg QD, 400 mg QD, 200 mg BID, or 600 mg BID. Each cohort had 10 subjects, with eight receiving active drug and two receiving matching placebo.
Twenty-one of the 40 patients were treatment-experienced and eight were coinfected with hepatitis C virus (HCV). Median baseline HIV-RNA levels ranged from 4.24 to 4.66 log10 copies/mL. Mean changes in HIV-RNA, after ten days of treatment, were -0.12 log10 in the placebo groups, -0.46 log10 among patients receiving 200 mg QD GW 874140, -1.23 log10 among patients receiving 200 mg BID, -1.03 log10 among patients receiving 400 mg QD, and -1.66 log10 among patients receiving 600 mg BID. The percentage of patients with a 1 log10 copies/mL drop or greater in viral load was 0% in the placebo group, 16.7% in the 200 mg QD group, 75% in the 200 mg BID group, 63% in the 400 mg QD group, and 100% in the 600 mg BID group. “Virologic suppression is similar to what we’re seeing with the other CCR5 antagonists,” Dr. Gulick said. “I think there’s a lot to look forward to with these three agents.”
The Concern of Coreceptor Switching
The three CCR5 antagonists are only expected to be active in people with R5-tropic virus. Studies have documented that, as HIV disease progresses, an increased percentage of patients have evidence of X4-tropic virus: HIV that is tropic for the CXCR4 receptor. Given that these agents do not work against X4-tropic virus, there’s some concern that R5 antagonists may result in the selection and proliferation of X4-tropic virus.
“What is the danger of this?” asked Dr. Gulick. “We really don’t now, but in people who have X4-tropic virus, there seems to be faster HIV disease progression. We’ve known this for more than ten years and there’s lingering concern about coreceptor switching in individuals receiving these drugs. In each of the CCR5 antagonist studies reported here, there has been evidence of coreceptor switching in one or two patients. Most, however, have switched back to R5-tropic virus when the compounds were withdrawn. Suffice it to say, the clinical implications aren’t yet clear.
AMD3100 and AMD070
AMD3100 and AMD070 are both CXCR4 antagonists from AnorMED Inc.. Because of toxicity concerns and limited efficacy, the development of AMD3100 was halted in May 2001. The company is now focused on AMD070, a compound that strongly inhibits viral infection by CXCR4-using virus—including virus using CXCR4 alone and/or virus using both CXCR4 and CCR5—in vitro. AMD070 is orally bioavailable in animals, shows additive or synergistic effects in vitro with other antiretroviral agents, and has yielded safe results in a recent Phase Ia dose-ranging study involving HIV-negative volunteers (Stone, 2004).
A Phase IIa study is being conducted by the Adult AIDS Clinical Trials Group (AACTG 5210).
| References | Top of page |