
The successful use of highly active antiretroviral therapy (HAART) can dramatically suppress human immunodeficiency virus (HIV)-1 viral replication and effect significant immune reconstitution.1–4 However, despite full access to antiretroviral agents, the emergence of antiretroviral-resistant HIV-1 strains and/or drug toxicities can derail effective treatment. A prospective study of patients in a New York City cohort with acute and early HIV-1 infection found the prevalence of transmitted resistance to at least one antiretroviral agent to be 24.1%.5 Consequently, the need to develop antiretroviral agents with novel mechanisms of action persists for the treatment of both antiretroviral-experienced and antiretroviral-naïve patients. In October 2007, the United States Food and Drug Administration (FDA) approved the first drug in the integrase-inhibitor class for the treatment of HIV-1 as part of combination antiretroviral therapy in treatment-experienced patients, adding to the available chemotherapeutic agents for the effective treatment of HIV/AIDS.
| HIV Integrase and Integration | Top of page |
Successful HIV-1 replication requires the use of 3 enzymes: reverse transcriptase, integrase, and protease. The HIV-1 life cycle initiates with viral entry into host immune cells that express surface CD4.6–9 After viral entry, HIV-1 reverse transcriptase converts its single-stranded RNA into double-stranded DNA (dsDNA), at which time integrase assembles in a stable complex with viral DNA—the pre-integration complex—and is chaperoned into the nucleus.10 Subsequent integration of HIV-1–complementary DNA (cDNA) into the host genome is a two-step process catalyzed by the HIV-1 integrase enzyme (Figure 1). Initially, 2 nucleotides are excised from the 3´ ends of the nascent HIV-1 DNA. This is followed by the irreversible, covalent insertion of HIV-1 viral genomic DNA into the host chromosome.11,12 While the HIV-1 virus is known to preferentially target sites within transcribed host genes for integration—so called “hot spots”—the factors underlying these preferences are not entirely clear.13,14 When integrase is inhibited, host enzymes circularize the viral cDNA, and 2-long terminal repeat (LTR) circles accumulate in the nucleus.15–18 Inhibiting integrase from performing its essential functions therefore blocks stable integration of HIV-1 DNA into the host genome and prohibits the establishment of viral latency within the host cell, preventing high-level HIV-1 replication and infection of new cells by competent virus.19
Figure 1. Integration of HIV-1–cDNA into host genome.
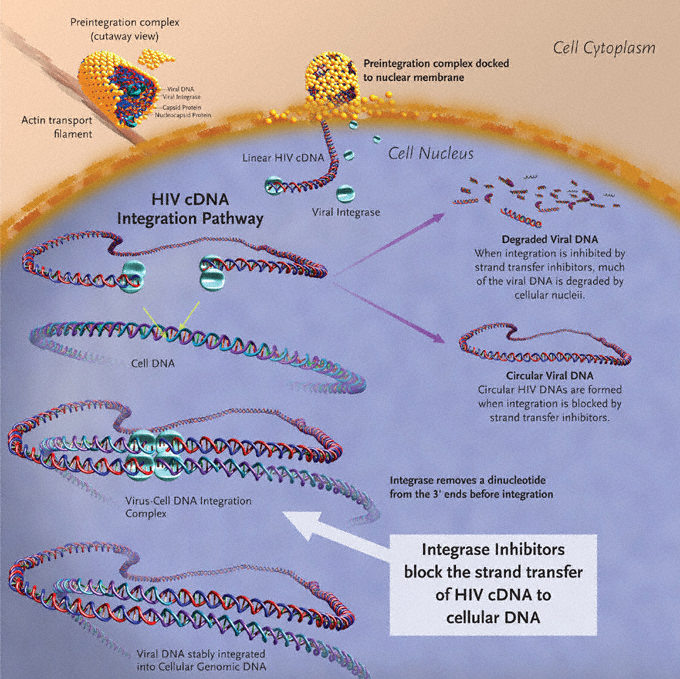
Image created by Louis E Henderson, PhD.
| Clinically Relevant Compounds | Top of page |
| Raltegravir (RAL, IsentressTM, formerly MK-0518) | Top of page |

Figure 2. The chemical structure of raltegravir.
Adapted with permission from Merck & Co., Inc.49
Raltegravir is a 1-N-alkyl-5-hydroxypyrimidinone. As such, it is a structural analogue of the di-keto acid class of compounds and shares their β-hydroxy-ketone structural motif (Figure 2).17,20,21 This structural motif possesses metal-chelating functions, and it is postulated that compounds bearing these functional groups interact with divalent metals within the active site of HIV-1 integrase.22–24 The insertion of HIV-1 viral genomic DNA into the host chromosome is a process often referred to as strand transfer.11,12 Raltegravir and its related molecules inhibit this latter step, and as a result are often referred to as “strand-transfer inhibitors.” The work of several authors provides an in-depth discussion of the chemical synthesis and screening of HIV-1 integrase inhibitors.25–27
Raltegravir has been shown to have a 50% inhibitory concentration (IC50) of approximately 10 nM.28 The results of 3 double-blind, randomized, placebo-controlled studies of raltegravir dosing demonstrate that raltegravir exhibits potent in-vitro activity and has an IC95 of 33 nM in 50% human serum.29 Raltegravir is active across diverse HIV-1 clinical isolates and has been shown to inhibit the in-vitro replication of HIV-2.28
| Elvitegravir (EVG, GS-9137, JTK-303) | Top of page |

Figure 3. The chemical structure of elvitegravir.
Adapted with permission from Gilead Sciences, Inc.
Elvitegravir is a dihydroquinoline carboxylic acid compound that, like raltegravir, exhibits the active integrase-inhibitor–conferring β-hydroxy-ketone structural motif (Figure 3). Also, like raltegravir, elvitegravir is a specific inhibitor of the strand-transfer step of HIV integration.30 This drug is active against HIV-1 and HIV-2, has an IC90 of 1.2 nM in peripheral blood mononuclear cells (PBMCs), and a serum-free antiviral IC50 of 0.2 nM. As expected, elvitegravir has also demonstrated activity against isolates resistant to nucleoside reverse-transcriptase inhibitors (NRTIs), non-nucleoside reverse-transcriptase inhibitors (NNRTIs), and protease inhibitors (PIs).31
| Clinical Trials | Top of page |
Monotherapy Proof-of-Concept Studies
In their randomized, double-blind, placebo-controlled study, the Protocol 004 study team established the in-vivo tolerability, pharmacokinetic profile, and antiviral activity of raltegravir. See Sidebar.
| Treatment-Experienced Patients | Top of page |
Protocol 005
The safety and efficacy of raltegravir in HIV-1–positive individuals with heavy antiretroviral treatment experience was investigated in a phase-IIb study by Grinsztejn et al (Protocol 005). This international, triple-blind trial randomized 179 HIV-1–positive participants, in a 1:1:1:1 ratio to 1 of 3 doses of raltegravir (200, 400, or 600 mg) or placebo twice daily, in combination with an optimized background regimen (OBR) of antiretrovirals (ARVs) chosen by the investigator. Patients were stratified by their degree of HIV-1 resistance to PIs as well as their use of the fusion inhibitor enfuvirtide in the OBR at study entry. Enrollment criteria included a CD4+ T-cell count >50 cells/mL, an HIV-1 RNA viral load >5000 copies/mL, documented genotypic and phenotypic resistance to at least 1 drug in each of 3 major classes of HIV-1 ARVs (NRTIs, NNRTIs, and PIs), and evidence of virologic failure at study entry.
Prior to enrollment, mean baseline HIV-1 viral load in the largely male cohorts was 4.7± 0.5 log10 copies/mL, and median treatment duration measured 9.9 years across study arms, with each group having taken a median of 4 antiretroviral combinations. Across treatment groups and placebo, the percent of patients using enfuvirtide as part of the OBR was 33%–38%; those with a phenotypic sensitivity score (PSS) of 0 to all available ARVs ranged from 40%–57%; and those with a PSS of 0 to PIs was 84%–98%.
A 24-week intention-to-treat (ITT) analysis revealed the mean change in viral load from baseline to be
- –0.35 (95% confidence interval [CI], –0.61 to –0.09) log10 copies/mL in the placebo group,
- –1.80 (95% CI, –2.10 to –1.50) log10 copies/mL in the 200-mg group,
- –1.87 (95% CI, –2.16 to –1.58) log10 copies/mL in the 400-mg group, and
- –1.84 (95% CI, –2.10 to –1.58) log10 copies/mL in the 600-mg group.
Compared with 14% in the placebo group, 57%–67% of individuals in the raltegravir groups achieved a viral load <50 copies/mL. Increases from baseline in CD4+ T-cell counts were also noted with raltegravir, particularly in those receiving the 400- and 600-mg doses. Furthermore, the likelihood of achieving a viral load <400 copies/mL at week 24 was increased from approximately 60% to 90% or more if enfuvirtide was included in the raltegravir-containing regimen.33 These results reveal the potential for raltegravir, when combined with other active agents in the OBR, to be of particular benefit to patients with extensive, or “heavy,” antiretroviral treatment histories.
BENCHMRK-1 and BENCHMRK-2
At present, clinical data for raltegravir is significantly more complete than for elvitegravir; raltegravir is the only drug in the integrase-inhibitor class to have entered phase-III clinical trials. Preliminary results presented in abstract from 2 large ongoing trials using raltegravir were reported at the 14th Annual Conference on Retroviruses and Opportunistic Infections (CROI).34,35 Entitled “Blocking integrase in treatment-Experienced patients with a Novel Compound against HIV-1: MeRcK” and commonly referred to as BENCHMRK-1 and BENCHMRK-2, the 16- and 24-week findings from these planned 156-week multicenter, randomized, placebo-controlled studies thus far extend and support previous results. These 2 studies differ primarily in the geographic distribution of enrolled subjects, with BENCHMRK-1 enrolling in Europe, Peru, and Asia/Pacific, and BENCHMRK-2 enrolling primarily in North and South America. Inclusion criteria include failure of antiretroviral therapy with an HIV-1 RNA level >1000 copies/mL and infection with HIV-1 resistant to at least 1 drug in each of 3 classes of oral ARVs (NRTIs, NNRTIs, PIs). The potential for drug interactions with concomitant antiviral agents, as well as considerations of variability in raltegravir levels between and within individuals, led to the selection of the 400-mg twice-daily dose of raltegravir for these studies. Patients were therefore randomized 2:1 to receive 400 mg of raltegravir twice daily or placebo, each dosed orally in combination with an OBR selected based on patients' prior treatment history and results from HIV-1 resistance testing. Six hundred and ninety-nine patients were enrolled in the 2 studies combined. In 19%–21% of subjects, enfuvirtide was included as part of the OBR. Darunavir or tipranavir were included in the OBR when warranted by resistance testing or patient history. Of note, darunavir/ritonavir was included as part of the OBR in 25%–27% of patients enrolled in BENCHMRK-1, as opposed to 45%–50% of patients enrolled in BENCHMRK-2; these differences may reflect differential access to these agents. The mean baseline CD4+ T-cell count and HIV-1 viral load was 156 cells/mm³ and 4.6 log10 copies/mL for the regimen that included raltegravir and 153 cells/mm³ and 4.5 log10 copies/mL for the placebo regimen. Enrollees had approximately 11 years of prior ARV therapy, and approximately 90% carried an AIDS diagnosis. In the 16-week primary analysis time point for BENCHMRK-1, approximately 61% of patients receiving raltegravir in addition to an OBR achieved HIV-1 RNA levels of <50 copies/mL compared to 33% of patients receiving placebo+OBR (P<0.001). Sixteen-week results from BENCHMRK-2 (Figure 4) were similar to those of BENCHMRK-1: HIV-1 RNA levels were <50 copies/mL in 62% of the raltegravir recipients and 36% of those receiving placebo.34,35
Figure 4. BENCHMRK-2: Plasma HIV RNA level <50 copies/mL (NC=F).
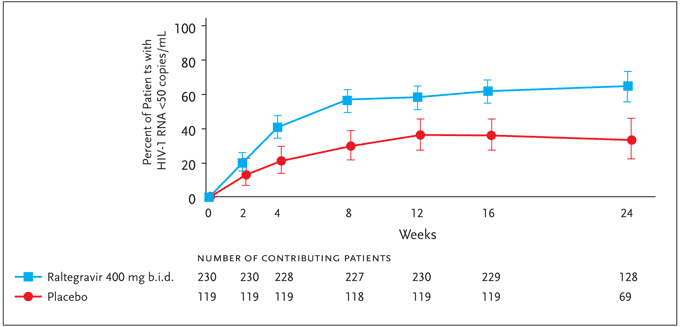
Adapted with permission from Merck & Co., Inc. Results of BENCHMRK-1 and-2, two phase-III studies evaluating the efficacy and safety of raltegravir, a novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus. 14th Conference on Retroviruses and Opportunistic Infections; February 25–28, 2007; Los Angeles, CA. Abstracts 105aLB and 105bLB. Copyright © 2007 Merck & Co., Inc., Whitehouse Station, NJ. All rights reserved.
Within the BENCHMRK studies, researchers also determined the percentage of patients achieving HIV-1 RNA levels of <400 copies/mL at week 16 within subgroups defined by study entry PSS and genotypic sensitivity scores (GSS). Not surprisingly, the likelihood of achieving an HIV-1 RNA level <400 copies/mL by week 16 was increased if raltegravir was combined with effective agents in the OBR. For example, by week 16, 57% (n=111) of patients with a GSS of 0 were able to achieve HIV-1 RNA levels <400 copies/mL with the addition of raltegravir to the OBR, as compared to 10% (n=63) in the placebo+OBR arm. However, in those with a GSS score of 2 or more, 89% (n=159) in the raltegravir+OBR arm were able to achieve viral suppression versus 71% in the placebo+OBR arm. Similar trends were noted in subgroups defined by baseline PSS. The first-time use of both enfuvirtide and darunavir as part of the OBR also greatly increased the participants’ chances for achieving virologic suppression by week 16. For those in whom both agents were used for the first time in the OBR, 98% (n=44) were able to achieve HIV-1 RNA levels <400 copies/mL (raltegravir+OBR) versus 87% (n=23) in the placebo+OBR arm. In those for whom neither enfuvirtide nor darunavir was used in the OBR, only 74% of the raltegravir+OBR arm were able to achieve viral suppression versus 29% in the placebo+OBR arm. Again, these results strongly support the combination of raltegravir with active agents in the OBR in the successful management of heavily treatment-experienced patients.
At 24-weeks, combined data from BENCHMRK-1 and -2 revealed that
- 75% of participants taking raltegravir+OBR achieved an HIV-1 viral load <400 copies/mL (versus 40% with placebo+OBR), and
- 63% in the raltegravir+OBR arm achieved an HIV-1 viral load of <50 copies/mL (versus 34% with placebo+ OBR [P<0.001 for both endpoints]).
On average, the raltegravir+OBR group gained 84 CD4+ T-cells from baseline, while the placebo group gained 37 CD4+ T-cells (P<0.001).36 Reductions in viral load observed at week 24 were sustained. After 48 weeks of therapy, 60%–65% of participants in the raltegravir arms of BENCHMRK-1 and -2 were found to have maintained HIV-1 viral suppression to <50 copies/mL.37–39
Gilead 0105
Twenty-four week clinical trials data from the Gilead study 0105 was presented in abstract at the 14th CROI.40 This is an ongoing, phase-II, randomized, dose-finding study in which the authors were partially blinded (to elvitegravir dose). This study was initially designed as a non-inferiority study of elvitegravir (boosted with 100 mg ritonavir) versus boosted PIs in treatment-experienced patients. Inclusion criteria for the 278 enrolled HIV-positive, predominantly male adults included HIV-1 RNA ≥1000 copies/mL, and ≥1 protease resistance mutation. At entry, mean HIV-1 RNA levels ranged from 4.54–4.71 log10 copies/mL, with mean CD4+ T-cell counts of 157–243 cells per mm3. Reflecting the heavy treatment experience of enrolled patients, 48%–51% of patients had a GSS of 0 for all NRTIs in the OBR in each arm of the study and approximately 11 PI resistance mutations in their HIV. Between 17% and 26% of patients were using enfuvirtide for the first time in the study.
Optimized background regimens consisted of nucleos(t)ide reverse-transcriptase inhibitors, with or without enfuvirtide when warranted by resistance studies or patient history. The use of protease inhibitors was initially prohibited in the elvitegravir arms, and NNRTIs were not allowed as part of the OBR in any arm of the study due to potential drug–drug interactions. Patients were stratified by enfuvirtide use in the OBR, and randomized 1:1:1:1 to receive 1 of 3 doses of elvitegravir (20 mg, 50 mg, or 125 mg), combined with 100 mg ritonavir for boosting purposes, once daily, versus a comparator FDA-approved ritonavir boosted PI (CPI/r). Of note, the (CPI/r) arm included 49% darunavir and 27% tipranavir use in the OBR.
Following review of the 8-week data, the Data and Safety Monitoring Board (DSMB) for the study recommended closing the 20-mg elvitegravir group due to a high rate of virologic failure. Patients in this arm were offered open-label elvitegravir at the 125-mg dose. In addition, as a result of new data indicating lack of drug–drug interactions, darunavir or tipranavir could now be added to ongoing elvitegravir arms if clinically warranted. Prior to week 16, only 4 patients added a PI to their regimens. As a result, week 16 was used as the latest time for comparison of enfuvirtide versus PI. By week 24, 15% of patients in the enfuvirtide 50- and 125-mg arms added a PI to their regimens. Using ITT at the 16-week study time point, the mean change from baseline in HIV-1 RNA log10 was
- –1.2 in the CPI group,
- –1.5 in the 50-mg elvitegravir group, and
- –1.7 in the 125-mg elvitegravir group.
Viral load reductions were similar after 24 weeks in the study. Of note, 37% of CPI patients switched to elvitegravir beginning at week 16. Additionally, as was previously mentioned, patients in the elvitegravir 20-mg arm were switched to 125-mg open-label elvitegravir beginning at study week 16. By the 24-week time point, 86%–99% of randomized patients had received at least 1 dose of study regimen. Utilizing the primary endpoint of time-weighted average change from baseline in HIV-1 RNA through 24 weeks, the 50-mg and 125-mg elvitegravir groups were deemed “non-inferior” to the CPI group. Furthermore, results from the 125-mg elvitegravir group were shown to be statistically superior to the CPI group at both 16 and 24 weeks (P=0.01 and 0.02 versus CPI/r respectively). Through week 24, approximately 11%–13% of patients in each arm discontinued in the study, with 3%–4% of those discontinuations attributed to issues of safety, tolerability, or efficacy. There was no increased evidence of grade-2, -3, or -4 adverse events in elvitegravir-exposed patients compared to the optimized background group.40
The authors also investigated the influence of activity of the OBR on the change from baseline in HIV-1 RNA levels with elvitegravir use at the 125-mg dose. Among patients entering the study with evidence of resistance to all of the available NRTIs and PIs (with no active drug in the OBR), the mean change from baseline in HIV-1 RNA was –0.7 log10 copies/mL at 24 weeks. However, if at least 1 active NRTI (or first use of enfuvirtide) was included in the OBR, the mean change from baseline in HIV-1 RNA was –2.1 log10 copies/mL at the same time point, a highly significant difference (P<.001). Of note, data from patients in the enfuvirtide 125-mg arm after addition of a PI were excluded from this analysis. At the 16-week time point, approximately 30% (n=63) of patients in the CPI group had viral loads <50 copies/mL, compared to 38% (n=71) of patients in the 50-mg elvitegravir group and 40% (n=73) of patients in the 125-mg elvitegravir group.40,41 As noted by the study authors, these results support the notion that the success of elvitegravir is highly dependent on the use of an OBR containing drugs to which the HIV isolate is sensitive.
| Naïve-patient studies | Top of page |
Protocol 004
In 2006, Markowitz et al published the results of Part II of Protocol 004, a combination antiretroviral therapy trial enrolling 201 treatment-naïve HIV-1–positive participants (including 30 from the raltegravir monotherapy trial [Part I]). In conjunction with tenofovir and lamivudine, all treated patients were randomized to receive 1 of 4 raltegravir doses (100, 200, 400, or 600 mg) twice daily versus efavirenz (600 mg/day). Inclusion criteria with respect to entry CD4+ T-cell count and HIV-1 RNA level were similar to that of Part I, as was the stratified randomization. (Those patients receiving placebo in Part I received efavirenz in Part II, while those treated with raltegravir in Part I retained the same drug dosage in Part II.) Characteristics were well balanced across treatment groups at baseline, with mean HIV-1 RNA levels ranging from 4.6–4.8 log10 copies/mL; mean CD4+ T-cell counts from 271–338 cells/mm3; and HIV-1 RNA levels >50,000 copies/mL in 55% of patients, and >100,000 copies/mL at baseline in 34% of patients.
Determinations of efficacy were based on a modified intention-to-treat (MITT) analysis with the primary endpoint being the proportion of patients achieving a plasma HIV-1 RNA level <400 copies/mL. Raltegravir was found to have a rapid and durable antiretroviral effect. By week 4, combination therapy with all study doses of raltegravir effected rapid and sustained reductions in plasma HIV-1 RNA levels, with at least 90% of patients reaching <400 copies/mL. At this time point, 60%–80% of patients in the raltegravir groups had suppressed their HIV-1 viral load to <50 copies/mL versus 25% of those treated with efavirenz (Figure 5). At the completion of 24 weeks (the primary time point for efficacy) and 48 weeks of therapy, differences between treatment groups diminished, with the plasma HIV-1 RNA level reduced to <50 copies/mL in up to 95% of all study subjects. These reductions in viral load were generally sustained through week 48 (Figure 5). The average increases in CD4+ T-cell counts were comparable across treatment groups at weeks 24 and 48. Reflecting the potency of raltegravir-based therapy, patients receiving raltegravir at any dose achieved HIV-1 RNA levels of <50 copies/mL statistically earlier than patients receiving efavirenz; the full clinical significance of this earlier virological suppression remains to be determined.
The majority of adverse events in Protocol 004 were graded mild (~85%) to moderate. Drug-related clinical adverse events were less frequent with raltegravir (48%) than efavirenz (71%). The most frequent raltegravir-related adverse events including nausea, dizziness, and headache. The incidence of serious adverse events was similar in patients receiving the raltegravir and efavirenz combination regimens (5% and 6% respectively), and the incidence of adverse events was not related to raltegravir dose. Not surprisingly, neuropsychiatric symptoms were less common with raltegravir than with efavirenz at weeks 8 and 48: 8% versus 21% and 13% versus 29%, respectively.
None of the serious adverse events in this study were considered to be drug related or led to treatment discontinuation. Additionally, grade-3 and -4 laboratory abnormalities were uncommon in this study. In patients receiving raltegravir, these included decreased absolute neutrophil count, transaminitis, or increased pancreatic enzymes. In addition, raltegravir was found to have a neutral effect on serum lipids: at week 48, the mean change from baseline in total cholesterol for raltegravir was –2.3 mg/dl versus +20.7 for efavirenz (P<0.001). Low-density lipoproteins and triglycerides were also relatively unchanged from baseline in the raltegravir groups but were increased in the efavirenz group.42
Figure 5. Efficacy differences of raltegravir versus efavirenz from Protocol 004 through week 48.
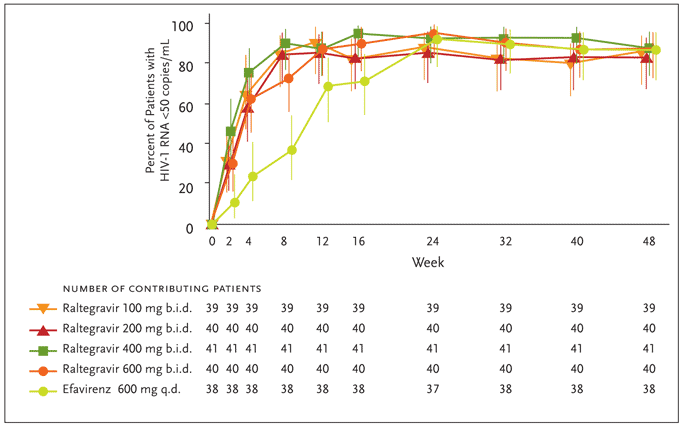
Differences versus efavirenz at Week 4 and 8 are statistically significant (P<.05).
From Markowitz M, Nguyen BY, Gotuzzo E, et al. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naïve patients with HIV-1 infection. J Acquir Immune Defic Syndr 2007;46(2):125-133. Reprinted with permission from Lippincott Williams & Wilkins; © 2007.
| Decay hypotheses | Top of page |
Decay Hypotheses
An intriguing aspect of the raltegravir-naïve 004 study was the documented accelerated HIV-1 RNA decay to <50 copies/mL in raltegravir-treated groups. See Sidebar Two.
| Resistance to Integrase Inhibitors | Top of page |
Virologic failures in clinical studies provide valuable information on raltegravir resistance. In the BENCHMRK studies, 41 patients in the raltegravir arm were deemed treatment failures. Mutations were described in 32 of these patients: N155H, Q148K/R/H, and infrequently Y143R/C.34,35 Data presented in abstract from the Merck Protocol 005 Study team are consistent with these findings. In their analysis of 35 patients with integrase mutations during virologic failure on raltegravir+OBR, two genetic pathways of mutations in the HIV-1 integrase gene were noted: N155H or Q148K/R/H. Both pathways were associated with raltegravir resistance, with the Q148 pathway of mutations resulting in measurably larger reductions in susceptibility (25-fold versus 10-fold for N155). The acquisition of N155 or Q148 mutations were found to result in cross-resistance to structurally diverse integrase inhibitors and the acquisition of additional mutations resulted in high-level resistance both in vitro and in vivo.43 Of note, these mutations point directly to the catalytic site of HIV-1 integrase.20 The cross-resistance exhibited by HIV-1 variants with N155 or Q148 mutations is therefore consistent with the supposition that integrase inhibition takes place by affecting binding of the common pharmacophore within the active catalytic site of HIV-1 integrase. Factors influencing selection of divergent pathways leading to resistance and their full clinical implications remain unclear.43
During in-vitro passage of wild-type HIV-1 in the presence of elvitegravir, 2 patterns of primary integrase resistance—T66I and E92Q—were found to be the most commonly selected. The E92Q mutation had the greatest effect on elvitegravir susceptibility, reducing it 33-fold, while the T66I mutation reduced susceptibility 15-fold. These assays reveal a high level of cross-resistance between elvitegravir and raltegravir, with the E92Q and T66I mutations reducing raltegravir susceptibility by 6.0-fold and 1.4-fold, respectively. Furthermore, these primary resistance mutations were often accompanied by secondary mutations in integrase. Specifically, H51Y, S147G, and E157Q were found to accompany the E92Q mutation, while F121Y (a mutation also associated with reduced raltegravir susceptibility), S153Y, and R263K accompanied the primary T66I mutation. These secondary mutations further reduced elvitegravir susceptibility.44,45
Much of the current information on elvitegravir resistance in vivo derives from an analysis of the integrase genotypes of viral isolates from protocol-defined virologic failures in the previously described phase-II, randomized, dose-finding study of elvitegravir (Gilead study 0105) in patients with heavy treatment experience. Integrase genotyping was performed on 28 of 30 patients with virologic failure in the ritonavir-boosted, elvitegravir 125-mg dosing arm by week 24. The most common integrase mutations developing in those patients were E92Q, E138K, Q148R/K/H, and N155H—each of which was observed in 39% of virologic failures; S147G (observed in 32%) and T66I/A/K (observed in 18%) complete the list of most commonly noted mutations.46 Despite their diverse structures, phenotypic analysis of HIV-1 from these patients also provided evidence for cross-resistance between the first-generation integrase inhibitors. Virus derived from virologic-failure patient samples demonstrated a mean elvitegravir fold change (FC) of greater than 151 (range 1.02–301) relative to the NL4-3 reference strain. These same samples demonstrated a raltegravir FC greater than 28-fold (range 0.78–256), consistent with reduced susceptibility across the first-generation integrase inhibitor class.46
| FDA Approval and Clinical Use | Top of page |
| Indications and Usage | Top of page |
On October 16, 2007, the FDA announced the approval of raltegravir for the treatment of HIV-1 infection as part of combination antiretroviral therapy in treatment-experienced patients with evidence of ongoing replication of HIV-1 strains resistant to multiple antiretroviral agents.47 For the treatment of patients with HIV-1 infection, the dosage of raltegravir is 400 mg administered orally, twice daily, with or without food. At present, raltegravir is the only drug in the integrase-inhibitor class approved for clinical use.
| Drug–Drug Interactions | Top of page |
In-vivo and in-vitro studies demonstrate that raltegravir is mainly eliminated via a uridine diphosphate glucuronosyltransferase 1 family, polypeptide A1 (UGT1A1)-mediated hepatic glucuronidation metabolic pathway.48 Glucuronosyltransferase inhibitors, as well as inducers of the enzyme, have the mechanistic potential to increase or decrease raltegravir concentrations, respectively. The elimination of raltegravir via the UGT1A1 metabolic pathway suggests caution be used in the coadministration of raltegravir with strong inducers of this pathway, such as rifampin, which could theoretically reduce raltegravir concentrations. The impact on UGT1A1 of other strong inducers of drug-metabolizing enzymes, such as phenytoin and phenobarbital, is unknown. Other less-strong inducers (eg, efavirenz, nevirapine, rifabutin, St. John’s wort) may be used with the recommended dose of raltegravir. Finally, unlike the PIs, raltegravir has no apparent effect on the cytochrome P450 3A4 (CYP3A4) system, and therefore has a low propensity to alter the pharmacokinetics of agents metabolized by CYP3A4.48 Additionally, raltegravir is not an inhibitor of UGT1A1, UDP glucuronosyltransferase 2 family, polypeptide B7 (UGT2B7), or P-glycoprotein-mediated transport.49
Much of the available data on drug–drug interactions with raltegravir is derived from pharmacokinetic studies presented in abstract at the 2006 Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). In a 14-day placebo-controlled study of 12 subjects receiving 400 mg raltegravir alone or in combination with 600 mg of efavirenz, the authors report a 21% reduction in C12hr and 36% reduction in both Cmax and AUC of raltegravir in the presence of the NNRTI. No significant decrease in Tmax or t1/2 was reported despite these differences.50 Similarly, the combination of 400 mg of raltegravir+100 mg ritonavir dosed twice daily had no significant impact on raltegravir pharmacokinetic parameters when compared to raltegravir administered alone.50 In healthy subjects, administration of the combination of 400 mg of raltegravir twice daily with standard-dose tenofovir for 4 days resulted in modest increases in raltegravir AUC (49%) and Cmax (64%). Cmin was unchanged. Conversely, the AUC was decreased by 10% and Cmin by 13% for tenofovir.51 These data do not suggest the need for dose adjustment with this combination. Additionally, the short-term addition of 400 mg raltegravir twice daily to steady-state tipranavir/ritonavir combination dose led to a 24% decrease in the tipranavir/ritonavir AUC. The Cmax decreased by 18%.52 Currently available information on the clinical occurrence of drug–drug interactions with raltegravir can be found in Table 1.
| Table 1. Reported Effects of Rifampin and HIV ARVs on the Pharmacokinetics of Raltegravir | |||||
| Ratio (90% CI) of Raltegravir Pharmacokinetic Parameters with/without Coadministered Drug; No Effect = 1.00 |
|||||
| Coadministered Drug Dose/Schedule | Raltegravir Dose/Schedule | n | Cmax | AUC | Cmin |
| Atazanavir 400 mg daily | 100 mg single dose | 10 | 1.53 (1.11, 2.12) | 1.72, 2.02) | 1.95 (1.30, 2.92) |
| Atazanavir 300 mg+ritonavir 100 mg daily | 400 mg single dose | 10 | 1.24 (0.87, 1.77) | 1.41 (1.12, 1.78) | 1.77 (1.39, 2.25) |
| Efavirenz 600 mg daily | 400 mg single dose | 9 | 0.64 (0.41, 0.98) | 0.64 (0.52, 0.80) | 0.79 (0.49, 1.28) |
| Rifampin 600 mg daily | 400 mg single dose | 9 | 0.62 (0.37, 1.04) | 0.60 (0.39, 0.91) | 0.39 (0.30, 0.51) |
| Ritonavir 100 mg twice daily | 400 mg single dose | 10 | 0.76 (0.55, 1.04) | 0.84 (0.70, 1.01) | 0.99 (0.70, 1.40) |
| Tenofovir 300 mg daily | 400 mg single dose | 9 | 1.64 (1.16, 2.32) | 1.49 (1.15, 1.94) | 1.03 (0.73, 1.45) |
| Tipranavir 500 mg+ritonavir 200 mg twice daily | 400 mg single dose | 15 (14 for Cmin) | 0.82 (0.46, 1.46) | 0.76 (0.49, 1.19) | 0.45 (0.31, 0.66) |
From: Isentress (raltegravir) [package insert]. Reprinted with permission from Merck & Co., Inc.49 |
|||||
| Adverse Reactions | Top of page |
The BENCHMRK studies provide the most complete information available thus far with regards to adverse events associated with the use of raltegravir. The most common adverse reactions reported in subjects in either the raltegravir or the placebo treatment group, regardless of causality, were nausea, headache, diarrhea, and pyrexia. While creatinine kinase elevations, myopathy, and rhabdomyolysis were observed in subjects receiving raltegravir, the true relationship of raltegravir to these events is currently unknown.34,35 As a result, the manufacturer recommends that raltegravir be used with caution in patients receiving concomitant medications that may place them at increased risk for these events.49 It should also be noted that although an early imbalance in the diagnosis of malignancies had been seen in the raltegravir-treated patients, this imbalance resolved with further follow-up. Post-approval surveillance will be needed to provide more definitive data on the rate, scope, and severity of integrase-inhibitor–related adverse events.
| Use in Special Populations | Top of page |
Although hepatic glucuronidation appears to be the major clearance mechanism of raltegravir in humans, clinical trials have revealed no important pharmacokinetic differences between healthy individuals and those with moderate hepatic impairment. At present, no dose adjustment appears necessary for those with mild to moderate disease. However, no recommendation can be made for the use of raltegravir in patients with severe hepatic impairment. No clinically important pharmacokinetic differences between those with severe renal impairment and healthy subjects have been reported when administered raltegravir. However, it should be noted that the extent to which raltegravir may be dialyzable is unknown.49
Raltegravir is currently classified as a category-C drug, and should therefore be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. There are no adequate clinical or pharmacokinetic studies of raltegravir in pregnant women. A similar situation exists for the pediatric patient, for whom no data from clinical trials exist.49
| Conclusions | Top of page |
As a new class of drug targeting the third essential enzyme for HIV replication (along with reverse transcriptase and protease), the integrase inhibitors are a welcome addition to the treatment armamentarium for HIV/AIDS in treatment-experienced patients failing available antiretroviral regimens.
Clinically relevant interactions with other available antiretroviral agents and long-term adverse effects and tolerability will have an impact on the future clinical value of the integrase inhibitors. Definition of the genetic barriers to integrase-inhibitor resistance, determinants of choice in the divergent pathways to resistance, and questions regarding cross-resistance across the class will need to be addressed. The integrase inhibitors should also be studied further as potential components in first-line HAART regimens based on available experimental data with raltegravir in combination with NRTIs. This would be particularly true for those in whom PI- or NNRTI-based therapy may be less than optimal. Finally, further safety, pharmacokinetic, and tolerability studies of raltegravir in special populations are warranted.
When considered as a whole, the promising efficacy and tolerability profile of the integrase inhibitors, absence of cross-resistance with other antiretroviral classes, and demonstrated synergism of the integrase inhibitors in combination with approved antiretroviral agents place them in a position to become important components of effective combination antiretroviral regimens in individuals living with HIV/AIDS.
| References | Top of page |