
Dr. Margolis began his October 2005 lecture with a crucial question. Two decades into the HIV/AIDS epidemic, what is to be done in the next decade? The answer, it seems, couldn’t be clearer: the evaluation of novel methods to prevent infection; the ongoing establishment of concrete treatment modalities to prevent disease in the infected; and, of particular interest to Dr. Margolis, continued momentum toward the eradication of infection.
The discussion of eradication is certainly not new to the pages of The PRN Notebook. Early treatment, intensified treatment regimens, and immune-based therapies to achieve this goal have all been pursued—with limited success—and discussed in detail over the past ten years. Just as it seemed as if the possibility of eradicating HIV was nothing more than a pipe dream, exciting new research has emerged—utilizing a truly novel approach and exploiting a very common compound—bringing the possibility of a cure to the forefront once again.
The compound in question is valproic acid, best known as Depakote, a tried and true treatment for bipolar disorder and epilepsy. The property of valproic acid exploited in the current approach is its known ability to inhibit histone deacetylases, enzymes that evidently maintain the integrated and latent HIV quiescent in the host genome. Although still in its infancy, the early work of Dr. Margolis and his colleagues has opened an exciting new avenue of research for the potential eradication of HIV. PRN was pleased to host Dr. Margolis at its October meeting to explain the theory behind, and the data supporting, continued evaluation of this approach.
| I. The Eradication Hypothesis Reborn | Top of page |
In 1995, experiments conducted at the Aaron Diamond AIDS Research Center (ADARC) and the University of Alabama independently established that HIV infection is a highly dynamic process, with a potential to produce more than 10 billion virions daily, even during the asymptomatic stage of disease (Ho, 1995; Wei, 1995). Employing antiretroviral therapy, these teams demonstrated that levels of HIV-RNA in plasma dropped by half approximately every 48 hours, signifying that enormous numbers of virions are produced by infected cells with relatively short life spans. Subsequent experiments documented the existence of a second, much slower phase of HIV clearance in plasma during therapy; this second phase was associated with the suppression of viral replication in long-lived cells in tissue. Simple mathematical modeling suggested that all cells harboring the virus would die off within about three years of maximally suppressive therapy, thereby raising the possibility of complete eradication of HIV from the human host.
The successful realization of this model with the currently available therapies depended on two key assumptions. First, the model presumed that all of the identified cellular populations had relatively the same short half-lives. And second, the estimate presumed that the suppression of viral replication achieved using combination antiretroviral therapy was, in fact, complete and total suppression of all HIV replication.
By November 1997, however, three research reports had laid to rest the first of these assumptions. The reports—representing a series of studies conducted at Johns Hopkins University School of Medicine, the University of California at San Diego, and the National Institutes of Health—confirmed the persistence of a viral reservoir consisting of latently infected, dormant memory CD4+ cells with integrated proviral DNA (Finzi, 1997; Chun, 1997; Wong, 1997). Given the estimated half-lives of the memory CD4+ cells range from six to 44 months, eradication would require anywhere from 10 to 60 years of chronic antiretroviral therapy.
Even this more sophisticated and biologically plausible reservoir-based hypothesis is dependent on the same key variable or fallacy—that of unconditional and total halt of viral replication with current antiretroviral therapy. Complete and absolute suppression is required in order to thoroughly snuff out latently infected cell populations and to prevent the reseeding of other long-lived cells. Studies completed over the past six years have unfortunately demonstrated that HIV replication does, in fact, persist even in those patients who consistently maintain undetectable viral loads with effective antiretroviral treatment (Zhang, 1999; Ramratnam, 2000). For eradication to be achieved, it is now clear that approaches to target and deplete latent infection within resting CD4+ cells are necessary.
One attempted approach has been the combination of intensive antiretroviral therapy with immune-based therapies—such as interleukin-2—to activate resting CD4+ cells. Forcibly activating these cells, the hypothesis held, would precipitate their death. And with the use of antiretroviral therapy, any released virus would be prevented from spreading and infecting adjacent cells.
While preliminary data from studies—see “Studies of HIV Latency: Implications for Treatment and Virus Eradication,” an article based on a presentation by Tae-Wook Chun, PhD, in the June 1999 issue of The PRN Notebook—were encouraging, final results (Chun, 1999) and additional research failed to demonstrate that this approach achieved viral eradication. A likely explanation for this, as was suggested in a paper published in 2000, is the fact that activation not only induces viral replication, it can increase the number of susceptible uninfected cells beyond the threshold that can be protected by antiretroviral therapy (Fraser, 2000).
What is needed, it seems, is an agent capable of inducing the expression of quiescent HIV, while simultaneously limiting any activation of CD4+ cells. One such approach being investigated by Dr. Margolis and his colleagues is the inhibition of histone deacetylase 1, an enzyme that appears to maintain latency of integrated HIV.
| Histone Deacetylation | Top of page |
Every latently infected CD4+ cell in the body harbors proviral HIV-DNA. The long terminal repeat (LTR) is a 100 base pair region at each end of proviral DNA and plays a vital role in viral transcription. LTR itself contains three regions: an upstream enhancer region, a core enhancer region, and a promoter region.
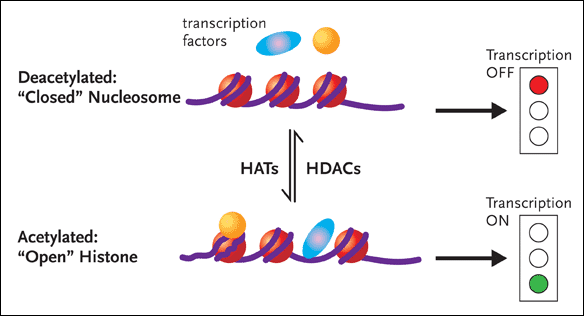
Figure 1. Chromatin RemodelingDNA is associated with proteins called histones to form the complex substance known as chromatin. Changes to the structure of chromatin have a profound influence on gene expression. If chromatin is condensed—closed—the transcription factors involved in gene expression cannot get to the DNA, and the genes will remain switched off (top half of figure). Conversely, if the chromatin is open, the genes can be switched on (bottom half of figure). Histone acetyltransferases (HATs) add an acetyl group to histones, resulting in an open chromatin structure. This, in turn, allows gene transcription to occur. There are also histone deacetylases (HDACs), enzymes that remove the acetyl groups from histones, resulting in less accessible chromatin structure. Dr. Margolis and his group hypothesize that inhibiting HDAC induces HAT and consequently promotes the outgrowth of HIV from latently infected CD4+ cells.
A number of cellular factors can act on any of the regions of LTR. An important example is the cellular transcription factor NFkB. It is believed that when NFkB is inactive, it cannot move from the cytoplasm into the cell’s nucleus where it binds to HIV’s LTR core enhancer region. The inhibitor protein IkBa is believed to be responsible for preventing NFkB from migrating to the nucleus.
If, however, NFkB is activated and succeeds in binding to the core enhancer region of LTR, changes in the local nucleosomal structure near the LTR promoter region occurs, with the enzymatic help of histone acetyltransferases (HATs). Here it is necessary to point out the DNA does not exist as coiled naked molecules in the cell. DNA is associated with proteins called histones to form the complex substance known as chromatin (see Figure 1). Changes to the structure of chromatin have a profound influence on gene expression. One simplified explanation holds that if chromatin is condensed—closed—the factors involved in gene expression cannot get to the DNA, and the genes will remain switched off. Conversely, if the chromatin is open, the genes can be switched on. HATs add an acetyl group to histones, resulting in an open chromatin structure. This, in turn, allows gene transcription to occur. There are also histone deacetylases (HDACs), enzymes that remove the acetyl groups from histones, resulting in less accessible chromatin structure.
The preliminary transcription initiated by NFkB activation of LTR results in a small burst of HIV gene expression. Among the first gene products to be produced are the HIV proteins tat and rev. Tat binds to the transactivation response (TAR) sequence at the beginning of the HIV-RNA in the nucleus and stimulates high-level HIV gene transcription and the accumulation of early HIV regulatory proteins, including rev. As rev accumulates to a critical level, it allows the production of structural viral proteins (e.g., env, gag, and pol) and viral enzymes ultimately promoting the formation of mature viral particles.
Research has determined that there are a number of cellular factors that are responsible for repressing viral transcription, at least in vitro. Of particular interest here are Yin Yang 1 (YY1) and late SV40 factor (LSF).
Synergy is the key with YY1 and LSF: both must be present and bind to LTR to repress transcription. The LTR binding site targeted by YY1 and LSF is known as the repressor complex sequence (RCS). Without LSF, YY1 has a difficult time binding to RCS. And without YY1, HDAC cannot be recruited to a key region of LTR known as nucleosome 1. If all goes smoothly—as it does in latently-infected CD4+ cells—LSF and YYI are both present. HDAC then interacts with the glycine/alanine-rich domain of YY1, resulting in removal of acetyl groups from the tails of core histones. Chromatin is then closed and HIV gene expression is inhibited.
| Inhibiting HDAC | Top of page |
Based on this intriguing process, researchers hypothesized that inhibiting HDAC would induce HAT and consequently promote the outgrowth of HIV from latently infected CD4+ cells. In 2001, it was determined that valproic acid (Depakote) is an inhibitor of HDAC, a finding that has resulted in great interest among researchers on the HIV eradication trail. In fact, the 2001 paper published in the Journal of Biological Chemistry proposes that the well-established teratogenicity of valproic acid is directly related to its inhibition of HDAC (Phiel, 2001). The paper also suggests that HDAC inhibition may explain valproic acid’s mechanism of action in the treatment of bipolar disorder.
In vitro studies completed in the mid-1990s demonstrated that valproic acid increased HIV gene expression and virus production in cell lines (Moog, 1996; Witvrouw, 1997). However, it wasn’t until 2004 that the world got its first glimpse at the effect of valproic acid on cells from HIV-positive patients. Published by Drs. Loyda Ylisastigui, Margolis, and their colleagues, these ex vivo studies indicated that valproic acid inhibition of HDAC allows for latent viral expression in resting CD4+ cells (Ylisastigui, 2004).
Using chromatin immunoprecipitation—a procedure used to determine whether a given protein is localized to a specific DNA sequence—these investigators found that valproic acid significantly increased acetylation at nucleosome 1 of LTR, the very region at which nucleosome acetylation is decreased by HDAC-1.
Lymphocytes were obtained by leukopheresis (the collection of leukocytes by a continuous flow cell separator) from five HIV-positive volunteers who had HIV-RNA levels below 50 copies/mL for at least 24 months. In the latently infected resting CD4+ cells, valproic acid induced outgrowth of HIV.
Cell surface phenotype analysis and de novo infection in the presence of valproic acid was also examined to explore the global affects of valproic acid on PBMCs in relation to HIV infection. In other words, the investigators wanted to see if valproic acid would increase de novo infection, much like IL-2 had in the earlier eradication studies. As was hoped, CD4+ cells exposed to valproic acid did not become activated or more permissive for viral replication.
Based on these results, the investigators concluded that valproic acid is a drug that may be used clinically to perturb the quiescent reservoir of HIV infection within resting CD4+ cells.
| A Proof-of-Concept Study | Top of page |
With his study team’s key in vitro study completed, Dr. Margolis and his colleagues then set out to test the ability of valproic acid to deplete HIV infection of resting CD4+ cells in vivo (Lehrman, 2005).
Conducted from July 2002 through February 2005, the study enrolled four HIV-infected individuals receiving antiretroviral therapy. These four patients had viral loads below 50 copies/mL for at least two years before entering the study. The clinical characteristics of the four patients, prior to and during the study, are reviewed in Table1.
| Table 1. Patients' Characteristics | ||||||
| Year Diagnosed | Treatment Regimen | Duration <50 copies/mL | CD4+ Count (%) | Serum Valproic Acid (mg/L) | ||
| Total | Free | |||||
| Patient 1 | 1995 | Tenofovir, abacavir, stavudine, amprenavir (with ritonavir) |
38 monts* | 1285 cells/mm3 (43%) |
65.1-83.1 |
5.8-6.7 |
| Patient 2 | 1999 | Didanosine, emtricitabine, efavirenz |
>43 months§ | 558 cells/mm3 (26%) |
52.4-89.6 |
3.0-9.8 |
| Patient 3 | 1985 | Tenofovir, abacavir, lamivudine, efavirenz, nelfinavir |
>241 months*§ | 350 cells/mm3 (18%) |
56.7-81.0 | 3.9-6.8 |
| Patient 4 | 1995 | Zidovudine, lamivudine, nevirapine |
>75 months§ |
372 cells/mm3 (35%) |
33.9-75.8 | 2.2-8.7 |
| *Patient 1 had a single measurement of 71 copies/mL HIV-RNA 96 weeks before initiating enfuvirtide; patient 3 had single measurements of 636 and 98 copies/mL HIV-RNA 12 and 72 weeks before initiating enfuvirtide. § Viremia suppressed before first visit to the clinic. Source: Lehrman, 2005. |
||||||
After two rounds of leukopheresis, the patients underwent treatment intensification with concomitant subcutaneous injections of enfuvirtide (Fuzeon). Four weeks after treatment intensification, oral valproic acid (500 to 750 mg BID) was initiated and continued for three months. The dose of valproic acid was adjusted to ensure plasma concentrations between 50 to 100 mg/L. After completion of valproic acid treatment, another round of leukopheresis was conducted. Dr. Margolis’ group estimated the number of resting CD4+ cells in infected units per billion (IUPB).
The four patients tolerated the treatment regimen and adhered well to therapy. All experienced enfuvirtide-related injection site reactions. One patient—Patient 4—had a history of antiretroviral-related anemia and experienced mild anemia during the last four weeks of treatment with valproic acid. A likely explanation for the anemia was the zidovudine component of his antiretroviral regimen. Valproic acid inhibits glucuronidation of zidovudine and increases its bioavailability, which theoretically increases the risk of zidovudine-induced anemia.
Dr. Margolis’ group did not uncover any evidence of immune activation in the four patients receiving valproic acid. There were no significant changes in concentrations of expression of known cell-surface markers of activation between the times of study entry, the completion of intensified antiretroviral therapy, and the completion of valproic acid treatment. There were no significant changes in the proportion of resting or activated in either the memory CD4+ or CD8+ cell populations. What’s more, lymphoproliferative assay responses to p24 antigen were low at study entry and remained unchanged throughout the study.
With respect to the effects of antiretroviral therapy and valproic acid on infection in resting CD4+ cells, an examination of the response of individual patients proved revealing. A snapshot of the frequency of infection in resting CD4+ cells before and after addition of enfuvirtide and valproic acid to the patients’ standard antiretroviral regimens is provided in Table 2.
Patient 1 had a single episode of transient viremia, possibly associated with an intercurrent infection, and also had low-level viremia that was unaffected by the addition of enfuvirtide. Resting cell IUPB did not decline impressively in this patient after intensified antiretroviral therapy and treatment with valproic acid.
Patient 2, however, had durable and complete suppression of detectable viremia, without any documented episodes of transient viremia, and without detectable viremia in a research assay capable of detecting a single copy of HIV-RNA/ml. Patient 2’s IUPB of resting CD4+ cells declined more than 84% by the end of the study. “He had no detectable plasma HIV-RNA throughout,” Dr. Margolis commented. “There was absolutely no virus recovered from resting cells. What we didn’t publish is that several months later, after he was back on his standard antiretroviral regimen, we conducted additional leukopheresis, for twice as long, and took 360 bulk cultures to the lab. We got only one positive culture. Clearly, there’s not much virus there.”
Patient 3 had two episodes of transient viremia during a state of immune activation prior to protocol therapy, along with one episode of transient viremia while on study treatment. Low-level viremia—detected using the single-copy assay—was detected throughout the study, despite the addition of enfuvirtide. Yet despite the low-level viremia, Patient 3’s IUPB of resting CD4+ cells decreased by 68% by the end of the study. “Patient 3 was one of the more interesting, if not the most interesting, patients in the study,” Dr. Margolis said. “He had detectable virus throughout the study: before, during, and after. This is an illustration of a point that’s been made by others. There is residual replication. So far, the intensified therapy that we’ve got doesn’t get it. There is also some compartmentalization of virus that’s hard to understand,” he added, referring to a blip in viremia detected in Patient 3’s semen while in the study. “We don’t know where the one episode of transient viremia [in peripheral blood] while on study—a viral load blip—came from. Did it start in the semen and then reach the blood, or was it in the blood and then reached the semen? We don’t know. These are definitely subjects for further study.”
The IUPB of patient 4’s resting CD4+ cells measured before protocol therapy was stable. Low-level viremia was detected before the addition of enfuvirtide, decreased after enfuvirtide therapy, and was less than 1 copy/mL one month after enfuvirtide was discontinued. After 12 weeks of valproic acid treatment, the observed reduction in IUPB was 72%.
Dr. Margolis summarized his team’s study as being an imperfect experiment. “We could only study four patients,” he said. “We can only say that this effect is seen as a result of therapy intensified with enfuvirtide and valproic acid together. We’re now doing the same study again, this time using just valproate; intensification with enfuvirtide will not be included. In our study, we only looked at the population of latently infected cells once after the start of valproic acid. This one point does not define a slope and we don’t know if this decline would flatten over time.”
Dr. Margolis also commented that there may be a population of latently infected cells that don’t respond to HDAC inhibition, or perhaps valproic acid is not potent enough. However, he noted that valproic acid likely achieves significant concentrations in lymphoid tissues. “It’s an anti-seizure drug. It achieves changes in gene expression in brain cells. Its volume of distribution is quite good. These are small molecules, which shouldn’t be a problem.
“Another valid criticism is that we only looked at circulating cells. Unfortunately, we can’t study the tissue compartments of these patients and collect enough cells safely to really count these rare viruses. These issues raise important questions for the future.”
Dr. Margolis also provided some possible explanations for the observed decreases in latently infected CD4+ cells. “We hope that the virus is being expressed and does what a resting cell is supposed to do next, which is proliferate and die.” As for what is mediating this effect, Dr. Margolis suggested that there are a few possibilities. “Clearly, valproate plays a role in HIV LTR chromatin remodeling. It’s also possible that valproate results in host gene modulation. This is why it raises patients’ seizure thresholds. Recently, there’s been a good argument that the acetylation of tat is important for tat activity and activation. So by inhibiting deacetylation, we’re basically increasing the activity of tat. Of course, it could be more than just one of these mechanisms.”
| Conclusion | Top of page |
In concluding his talk, Dr. Margolis reviewed some of the tasks ahead. “To move this field forward, we really need better assays than the terribly labor-intensive leukopheresis assay,” he said. “Ideally, a model system for latency or eradication should be developed.” He also mentioned that there are other HDAC inhibitors being explored—not necessarily for HIV infection—including several being evaluated in cancer trials. “Would these other HDAC inhibitors be too potent and toxic? Or would they be better than valproic acid? We don’t know.”
Dr. Margolis also noted that there are approaches other than targeting HDAC that might facilitate this same mechanism. “Critical is the characterization of persistent replication and finding out why it’s happening, where it’s happening, and how to avoid it,” he commented. “We want to be able to test new antiretrovirals, as well as vaccines, because all of these patients that have suppressed viremia have lost anti-HIV immune responses. If we are to clear this chronic infection, we will likely need the help of the immune system. Ideally, in the future, we will have strategies that turn on a viral expression program, and perhaps even a cell expression program, that results in low-level virus production or no virus production, along with death of the infected cell without too many toxic effects on the human host.”
| References | Top of page |